



1. Entendimento inicial
Nesta fase, precisamos entender alguns conceitos e terminologias, para evitar cometer erros diante de nossos idosos, como:
P: Qual é a diferença entre RT-PCR, qPCR, PCR em tempo real e RT-PCR em tempo real?
Resposta: RT-PCR é PCR de transcrição reversa(PCR de transcrição reversa, RT-PCR), que é uma variante amplamente utilizada da reação em cadeia da polimerase (PCR).Na RT-PCR, uma fita de RNA é transcrita reversa em DNA complementar, que é então usado como modelo para amplificação de DNA por PCR.
PCR em tempo real e qPCR(Quantitative Real-time-PCR) são a mesma coisa, ambos são PCR quantitativo em tempo real, o que significa que cada ciclo de PCR possui registros de dados em tempo real, de modo que o número de modelos iniciais pode ser ajustado para uma análise precisa.
Embora tanto a PCR em tempo real (PCR quantitativa fluorescente em tempo real) quanto a PCR de transcrição reversa (PCR de transcrição reversa) pareçam ser abreviadas como RT-PCR, a convenção internacional é: RT-PCR refere-se especificamente à transcrição reversaPCR , PCR em tempo real é geralmente abreviado como qPCR (PCR quantitativo em tempo real).
E RT-PCR em tempo real (RT-qPCR), é o PCR de transcrição reversa combinado com a tecnologia quantitativa fluorescente: primeiro obtenha cDNA (RT) da transcrição reversa do RNA e, em seguida, use PCR em tempo real para análise quantitativa (qPCR).A maioria dos laboratórios faz RT-qPCR, ou seja, pesquisa sobre down-regulation da expressão do RNA, então o qPCR que todo mundo fala no laboratório na verdade se refere ao RT-qPCR, mas não se esqueça que ainda existem muitos testes de DNA em aplicações clínicas.Análise quantitativa, como detecção de HBV do vírus da hepatite B.
Pergunta: Depois de ler muito PCR quantitativo fluorescente, por que o fragmento amplificado deve ser controlado na faixa de 80-300bp?
Responder: O comprimento de cada sequência de gene é diferente, alguns são vários kb, alguns são centenas de bp, mas só precisamos exigir que o comprimento do produto seja 80-300bp ao projetar primers, muito curtos ou muito longos não são adequados para detecção de PCR quantitativo fluorescente.O fragmento do produto é muito curto para ser distinguido do primer-dímero.O comprimento do primer-dímero é de cerca de 30-40bp, e é difícil distinguir se é um primer-dímero ou um produto se for menor que 80bp.Se o fragmento do produto for muito longo, excedendo 300 pb, ele levará facilmente a uma baixa eficiência de amplificação e não poderá detectar efetivamente a quantidade do gene.
Por exemplo, quando você conta quantas pessoas estão em uma sala de aula, você só precisa contar quantas bocas existem.O mesmo é verdade quando você detecta genes, você só precisa detectar uma certa sequência de um gene para representar A sequência inteira serve.Se você quiser contar pessoas, precisará contar bocas e narizes, orelhas e óculos, e é fácil cometer erros.
Para expandir, na pesquisa biológica, há muitos casos de pesquisa de ponto a área, porque a sequência gênica de qualquer espécie é muito longa, é desnecessário e impossível medir todos os fragmentos, como o sequenciamento bacteriano 16S, que é realizar ensaios de sequência conservativa de bactérias para inferir o número de uma determinada população de bactérias.
P: Qual é o comprimento ideal para o design do primer qPCR?
Responder: De um modo geral, o comprimento do primer é de cerca de 20-24bp, o que é melhor.Obviamente, devemos prestar atenção ao valor TM do primer ao projetar o primer, porque isso está relacionado à temperatura ideal de recozimento.Depois de muitos experimentos, provou-se que 60°C é um valor de TM melhor.Se a temperatura de recozimento for muito baixa, isso levará facilmente a uma amplificação não específica.Se a temperatura de recozimento for muito alta, a eficiência de amplificação será relativamente baixa, o pico da curva de amplificação começará mais tarde e o valor de CT será atrasado.
P: Qual a diferença entre o método do corante e o método da sonda?
Resposta: Método de tinturaAlguns corantes fluorescentes, como SYBR Green Ⅰ, PicoGreen, BEBO, etc., não emitem luz por si mesmos, mas emitirão fluorescência após a ligação ao sulco menor do DNA de fita dupla.Portanto, no início da reação de PCR, a máquina não consegue detectar o sinal fluorescente.Quando a reação atinge o estágio de recozimento-extensão, a fita dupla é aberta e uma nova fita é sintetizada sob a ação da DNA polimerase, e a molécula fluorescente se liga ao sulco menor do dsDNA.À medida que o número de ciclos de PCR aumenta, mais e mais corantes são combinados com o DNA de fita dupla, e o sinal fluorescente também é continuamente aprimorado.O método de corante é usado principalmente em pesquisas científicas.
PS: Cuidado ao fazer o experimento, o corante tem que ser combinado com DNA humano, cuidado para transformá-lo em uma pessoa fluorescente.
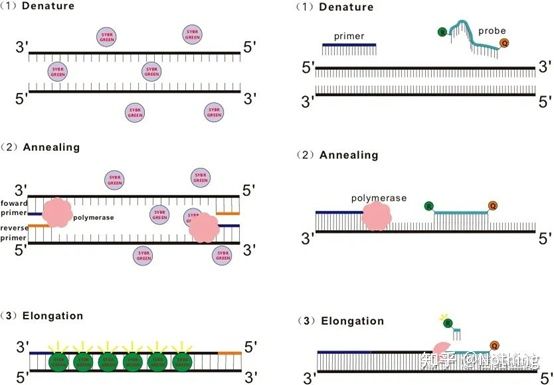
Método Dye (esquerda) Método Probe (direita)
PS: Cuidado ao fazer o experimento, o corante tem que ser combinado com DNA humano, cuidado para transformá-lo em uma pessoa fluorescente.
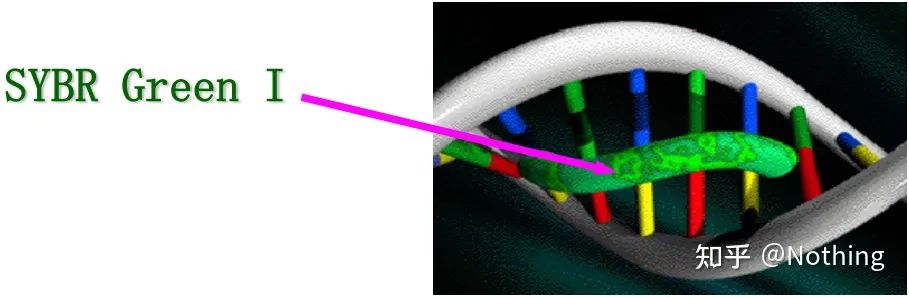
SYBR Green Ⅰ se liga ao sulco menor do DNA
Método de sondagemA sonda Taqman é a sonda de hidrólise mais comumente usada.Existe um grupo fluorescente na extremidade 5' da sonda, geralmente FAM, e a própria sonda é uma sequência complementar ao gene alvo.Há um grupo de extinção fluorescente na extremidade 3'.De acordo com o princípio da transferência de energia de ressonância de fluorescência (transferência de energia de ressonância de Förster, FRET), quando o grupo fluorescente repórter (molécula fluorescente doadora) e o grupo fluorescente de extinção (molécula fluorescente receptora) são excitadosPortanto, no início da reação de PCR, quando a sonda estiver livre e intacta no sistema, o grupo fluorescente repórter não emitirá fluorescência.Ao recozimento, o primer e a sonda se ligam ao molde.Durante o estágio de extensão, a polimerase sintetiza continuamente novas cadeias.A DNA polimerase tem atividade exonuclease 5'-3'.Ao atingir a sonda, a DNA polimerase irá hidrolisar a sonda do molde, separar o grupo fluorescente repórter do grupo fluorescente supressor e liberar o sinal fluorescente.Como existe uma relação de um para um entre a sonda e o modelo, o método da sonda é superior ao método do corante em termos de precisão e sensibilidade do teste.O método da sonda é usado principalmente no diagnóstico.
P: O que é quantificação absoluta?O que é Quantificação Relativa?
Responder: A quantificação absoluta refere-se ao cálculo do número inicial de cópias da amostra a ser testada por qPCR, como quantos vírus HBV existem em 1ml de sangue.O resultado obtido pela quantificação relativa é a mudança na quantidade do gene alvo em uma amostra específica em relação a outra amostra de referência, e a expressão do gene é regulada para cima ou para baixo.
P: A quantidade de extração de RNA, a eficiência da transcrição reversa e a eficiência da amplificação afetarão os resultados experimentais?
P: O armazenamento de amostras, reagentes de extração, reagentes de transcrição reversa e consumíveis de transmissão de luz afetarão os resultados experimentais?
Q: Que método pode corrigir os dados experimentais?
Com relação a esses problemas, iremos descrevê-los em detalhes nas seções avançadas e avançadas abaixo.
2. Conhecimento avançado
No que diz respeito ao PCR quantitativo fluorescente em tempo real, devemos reconhecer a realidade de que milhares de trabalhos de pesquisa científica são publicados todos os anos, entre os quais a tecnologia de PCR quantitativo fluorescente não é um número pequeno.
Se não houver um padrão comum para medir o experimento de PCR quantitativo fluorescente, os resultados podem variar amplamente.Para o mesmo gene da mesma espécie, com o mesmo método de processamento, os resultados da detecção também variam muito e será difícil para os retardatários repetir os mesmos resultados.Você Ninguém sabe o que é certo e o que é errado.
Isso significa que a PCR quantitativa fluorescente é uma tecnologia fraudulenta ou não confiável?Não, é porque o PCR quantitativo fluorescente é mais sensível e mais preciso, e uma pequena operação errada produzirá resultados completamente opostos.Uma pequena perda está a mil milhas de distância.O autor do artigo pode ser repetidamente torturado pelos revisores.Ao mesmo tempo, os revisores da revista também têm dificuldade em escolher entre diferentes resultados experimentais.
Em suma, apontando para uma falta de consenso em experimentos de PCR em tempo real.Para este fim, cientistas seniores da indústria começaram a formular padrões,exigindo que os contribuidores forneçam alguns detalhes experimentais e de processamento de dados necessários (incluindo dados necessários) no artigo para atender a esses padrões .
Os revisores podem julgar a qualidade do experimento lendo esses detalhes;futuros leitores também podem usar isso para repetir o experimento ou melhorar o experimento.Então, os resultados experimentais obtidos dessa maneira são cheios de informações, de alta qualidade e utilizáveis.
MIBBI (Informações Mínimas para Investigações Biológicas e Biomédicas -http://www.mibbi.org) passou a existir.MIBBI é um projeto que fornece padrões para experimentos.É publicado na natureza.Este projeto visa vários experimentos biológicos, incluindo biologia celular, Microarray, qPCR que vamos discutir agora, etc., e prevê cada tipo de experimento ao enviar manuscritos.Essa informação deve ser fornecida em todos os momentos.
No projeto MIBBI, existem dois artigos relacionados com PCR quantitativo fluorescente, nomeadamente:
·RDML (Real-Time PCR Data Markup Language) – uma linguagem estruturada e um guia de relatórios para dados PCR quantitativos em tempo real;
·MIQE (Informações Mínimas para Publicação de Experimentos de PCR Quantitativo em Tempo Real) – informações mínimas para publicação de artigos sobre experimentos de PCR quantitativo em tempo real.
Primeiro, vamos falar sobre RDML, a especificação de terminologia.
Se não houver uma definição padrão para tudo, é impossível continuar a discussão, por isso a explicação dos termos é tão importante no exame.
A terminologia usada no experimento de PCR quantitativo fluorescente inclui o seguinte conteúdo.A QIAGEN fez o melhor resumo para nós.Os seguintes estão todos secosbens .
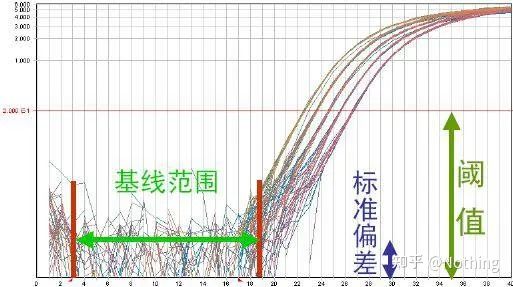
curva de amplificação
A curva de amplificação refere-se à curva feita durante o processo de PCR, com o número do ciclo como abscissa e a intensidade de fluorescência em tempo real durante a reação como ordenada.
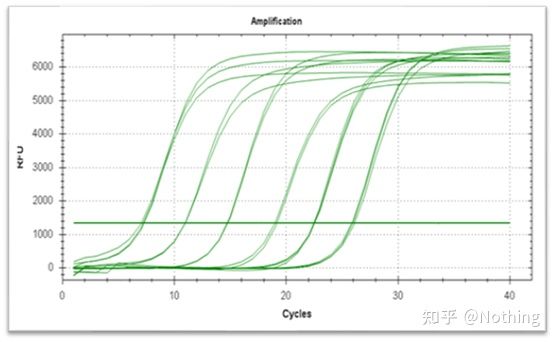
Uma curva de amplificação excelente deve ter as seguintes características: a linha de base é plana ou ligeiramente diminuída e não há tendência óbvia de aumento;o ponto de inflexão da curva é claro e a inclinação da fase exponencial é proporcional à eficiência de amplificação.Quanto maior a inclinação, maior a eficiência de amplificação;a curva de amplificação geral O paralelismo é bom, indicando que a eficiência de amplificação de cada tubo é semelhante;a fase exponencial da curva de amplificação de amostras de baixa concentração é óbvia.
Linha de base (linha de base)
A linha de base é o nível de ruído do ciclo inicial, geralmente medido entre o 3º e o 15º ciclo, pois o aumento do valor da fluorescência causado pelo produto de amplificação não pode ser detectado durante este período.O número de ciclos usados para calcular a linha de base pode ser variado e pode precisar ser reduzido se grandes quantidades de molde forem usadas ou se o nível de expressão do gene alvo for alto.
A definição da linha de base requer a visualização dos dados de fluorescência da curva de amplificação de linearidade.A linha de base é definida de modo que o crescimento da curva de amplificação comece com um número de ciclo maior que o número superior do ciclo da linha de base.As linhas de base precisam ser definidas individualmente para cada sequência alvo.Os valores médios de fluorescência detectados nos primeiros ciclos precisam ser subtraídos dos valores de fluorescência obtidos nos produtos amplificados.As versões mais recentes de vários softwares de PCR em tempo real permitem a otimização automática das configurações de linha de base para amostras individuais.
Durante os primeiros ciclos da reação de amplificação por PCR, o sinal de fluorescência não muda muito.Aproximar-se de uma linha reta é chamado de linha de base, mas se olharmos atentamente para os primeiros ciclos, veremos que dentro da linha de base está o que está acontecendo na figura abaixo.
Antecedentes Antecedentes refere-se a
o valor de fluorescência não específica na reação.Por exemplo: extinção de fluorescência ineficiente;ou um grande número de modelos de DNA de fita dupla devido ao uso de SYBR Green.Os componentes de fundo do sinal são matematicamente removidos pelo algoritmo do software Real-Time PCR.
Sinal do repórter
O sinal do repórter refere-se ao sinal fluorescente gerado por SYBR Green ou sondas específicas de sequência marcadas com fluorescência durante a PCR em tempo real.
Sinal Reporter Normalizado (RN)
RN refere-se à intensidade de fluorescência do corante repórter dividida pela intensidade de fluorescência do corante de referência passiva medida em cada ciclo.
Corante de referência passiva
Em alguns PCRs em tempo real,o corante fluorescente ROX é usado como referência interna para normalizar o sinal fluorescente.Ele corrige as variações devido à pipetagem imprecisa, posição do poço e flutuações de fluorescência poço a poço.

O limiar de fluorescência (limiar)
foi ajustado acima do valor de fundo e significativamente abaixo do valor de platô da curva de amplificação.Ele deve estar na região linear da curva de amplificação, representando o intervalo log-linear de detecção de PCR.Os limiares devem ser definidos na visualização da curva de amplificação logarítmica para que a fase linear logarítmica da PCR seja facilmente identificável.Se houver vários genes-alvo na PCR em tempo real, o limite deve ser definido para cada alvo.Geralmente, o sinal de fluorescência dos primeiros 15 ciclos da reação de PCR é usado como sinal de fundo de fluorescência, e o limite de fluorescência é 10 vezes o desvio padrão do sinal de fluorescência dos primeiros 3 a 15 ciclos de PCR, e o limite de fluorescência é definido na fase exponencial da amplificação por PCR.Em geral, cada instrumento tem seu limite de fluorescência definido antes do uso.
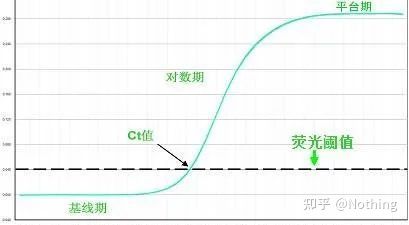
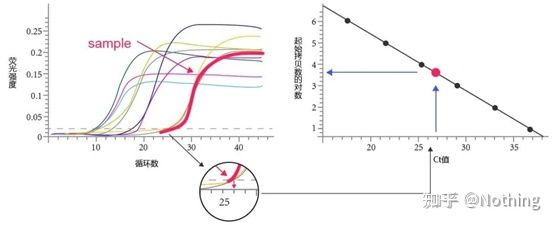
Limiar do Ciclo (CT) ou Ponto de Cruzamento (CP)
O ciclo no qual a curva de amplificação cruza o limiar (ou seja, o ponto no qual a detecção de fluorescência aumenta significativamente).CT pode ser uma fração e a quantidade de gabarito inicial pode ser calculada.O valor de CT representa o número de ciclos experimentados quando o sinal fluorescente em cada tubo de reação de PCR atinge o limite definido.Existe uma relação linear entre o valor CT de cada modelo e o logaritmo do número de cópias inicial do modelo, omaior o número de cópias inicial, menor o valor de CT e vice-versa.Uma curva padrão pode ser feita usando um padrão com número de cópias inicial conhecido, em que a abcissa representa o valor de CT e a ordenada representa o logaritmo do número de cópias inicial.Portanto, desde que o valor CT da amostra desconhecida seja obtido, o número inicial de cópias da amostra pode ser calculado a partir da curva padrão.
valor ΔCT
O valor ΔCT descrevea diferença entre o gene alvo e o valor CT do gene de referência endógeno correspondente, como um gene de manutenção, e é usado para normalizar a quantidade de modelo usado:
⇒ΔCT = CT (gene alvo) – CT (gene endógeno de referência)
Valor ΔΔCT
O valor ΔΔCT descreve a diferença entre o valor médio de ΔΔCT de uma amostra de interesse (por exemplo, células estimuladas) e o valor médio de ΔΔCT de uma amostra de referência (por exemplo, células não estimuladas).A amostra de referência também é chamada de amostra de calibração e todas as outras amostras são normalizadas para isso para quantificação relativa:
⇒ΔΔCT = média ΔCT (amostra de interesse) – média ΔCT (amostra de referência)
Genes endógenos de referência (genes endógenos de referência)
Os níveis de expressão de genes de referência endógenos, como genes de manutenção (housekeeping genes), não diferem entre as amostras.A comparação dos valores CT do gene de referência com o gene alvo permite que o nível de expressão do gene alvo seja normalizado para a quantidade de RNA ou cDNA de entrada (consulte a seção sobre valores ΔCT acima).
Genes de referência interna corrigem parapossível degradação do RNA ou presença de inibidores enzimáticos nas amostras de RNA, bem como variações no conteúdo do RNA, eficiência da transcrição reversa, recuperação de ácido nucléico e manuseio da amostra.Para selecionar o(s) gene(s) de referência ideal(is), modificamos o algoritmo para permitir a seleção da referência ideal dependente da configuração experimental.
Controle interno
Uma sequência de controle que é amplificada na mesma reação que a sequência alvo e sondada com uma sonda diferente (ou seja, realizando PCR duplex).Os controles internos são frequentemente usados para descartar amplificações com falha, como quando a sequência alvo não é detectada.
Amostra de calibração
Uma amostra de referência (por exemplo, RNA purificado de uma linha celular ou tecido) usada na quantificação relativa para comparar todas as outras amostras para determinar o nível de expressão relativa de um gene.A amostra de calibração pode ser qualquer amostra, mas geralmente é um controle (por exemplo, uma amostra não tratada ou uma amostra do tempo zero do experimento).
Controles positivos
usar reações de controle comquantidades conhecidas de modelo.Controles positivos são frequentemente usados para verificar se um conjunto de primer ou conjunto de primer-sonda está funcionando corretamente e se a reação está configurada corretamente.
Sem controle de modelo (NTC)
Uma reação de controle que contém todos os componentes necessários da reação de amplificação, exceto o molde, que geralmente é substituído por água.O uso de NTC pode encontrar a contaminação causada por contaminação de reagente ou DNA estranho, garantindo assim a autenticidade e confiabilidade dos dados de detecção.A amplificação do controle NTC indica contaminação.
Sem controle RT (NRT)
O processo de extração de RNA pode conter DNA genômico residual, que é extremamente prejudicial e é o culpado que afeta a qualidade dos dados e o inimigo natural do qPCR; portanto, ao projetar experimentos, ele deve ser projetado para apenas amplificar a detecção de RNA.Existem duas maneiras, uma é projetar primers nos íntrons, a outra é remover completamente o DNA, qual é o melhor, o que será discutido mais adiante.O controle NTR é um espelho mágico para detectar a poluição do DNA.Se houver amplificação, significa que há poluição.
Padrões
Padrões são amostras de concentração conhecida ou número de cópias que são usadas para construir uma curva padrão.Para garantir a estabilidade do padrão, o fragmento do gene é geralmente clonado no plasmídeo e usado como padrão.
A curva padrão
é geralmente diluído em pelo menos 5 gradientes de concentração com o produto padrão de acordo com a taxa de duplicação, e 5 pontos são desenhados nas coordenadas do valor CT e número de cópias, e os pontos são conectados para formar uma linha para gerar uma curva padrão.Para cada curva padrão, sua validade precisa ser verificada.O valor da inclinação cai entre –3,3 e –3,8 e cada concentração é realizada em triplicado.Pontos que são significativamente diferentes de outros pontos devem ser descartados.O valor CT da amostra a ser testada é trazido para a curva padrão e o nível de expressão da amostra a ser testada pode ser calculado.
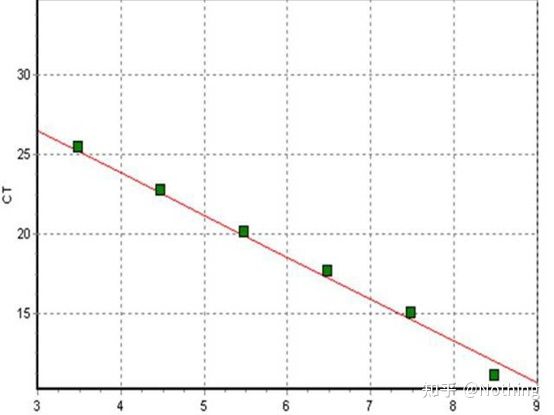
O valor CT da amostra a ser testada é trazido para a curva padrão e o número de cópias inicial da amostra a ser testada pode ser calculado.
Eficiência e Inclinação
A inclinação da curva padrão representa a eficiência da PCR em tempo real.
·Uma inclinação de -3,322 indica que a eficiência da amplificação de PCR é 1, ou 100% eficiente, e a quantidade de produto de PCR dobra a cada ciclo.
·Uma inclinação menor que -3,322 (por exemplo, -3,8) indica uma eficiência de PCR
·Uma inclinação maior que –3,322 (por exemplo, –3,0) indica que a eficiência da PCR parece ser maior que 100%, o que é curioso, como um ciclo de PCR poderia gerar mais que o dobro do produto amplificado?Essa situação ocorre na fase não linear da reação de PCR, ou seja, há uma grande quantidade de amplificação não específica.
curva de fusão
Depois que a amplificação qPCR é concluída, o produto de PCR é aquecido.À medida que a temperatura aumenta, o produto de amplificação de cadeia dupla derrete gradualmente, resultando numa diminuição da intensidade da fluorescência.Quando uma certa temperatura (Tm) é atingida, um grande número de produtos irá derreter.A fluorescência cai drasticamente.Diferentes produtos de PCR têm diferentes valores de Tm e diferentes temperaturas de fusão, de modo que a especificidade da PCR pode ser identificada.
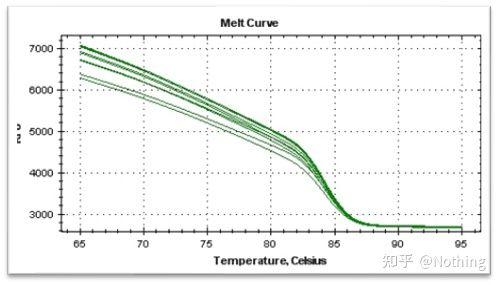
Curva de fusão (curva derivada)
A curva de fusão é derivada para formar um mapa de pico, que pode exibir de forma mais intuitiva a situação dos fragmentos de produtos de PCR.Uma vez que a temperatura de fusão é o valor Tm do fragmento de DNA, alguns parâmetros que afetam o valor Tm do fragmento de DNA podem ser julgados, como tamanho do fragmento, conteúdo GC, etc. De um modo geral, de acordo com nossos princípios de design de primer,o comprimento do produto amplificado está na faixa de 80-300 pb, então a temperatura de fusão deve estar entre 80°C e 90°C.
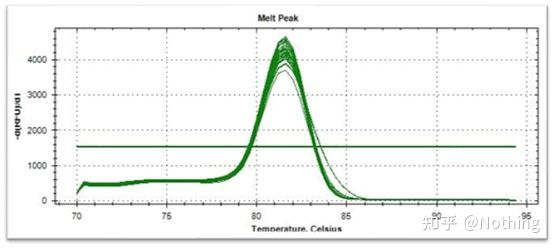
Interpretação da curva de fusão: Se o único pico principal aparecer entre 80°C-90°C, significa que a PCR quantitativa fluorescente é perfeita;se o pico principal aparecer entre 80°C-90°C e picos diversos aparecerem abaixo de 80°C, o dímero do iniciador é basicamente considerado.Você pode tentar aumentar a temperatura de recozimento para resolvê-lo;se o pico principal aparecer entre 80°C-90°C, e o pico miscelâneo aparecer novamente quando a temperatura subir, considera-se basicamente que há contaminação de DNA, e o DNA precisa ser removido no estágio inicial do experimento.
Claro, ainda existem algumas situações anormais, que serão discriminadas uma a uma abaixo.
3. Conhecimento avançado
Para fazer qPCR, devo dizer MIQE,Informação Mínimapara Publicação dequantitativoPCR em tempo realExperimentos — as informações mínimas para publicar artigos sobre PCR quantitativo em tempo realexperiências .A fim de simplificar a compreensão de todos, simplificaremos o conteúdo principal.
Você pode pesquisar o texto original do MIQE na Internet, e o mais importante é que ele estipula olista de verificação de dados que precisa ser fornecida ao publicar um artigo .
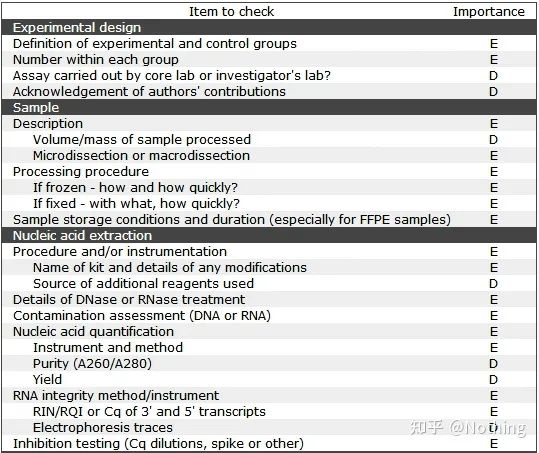
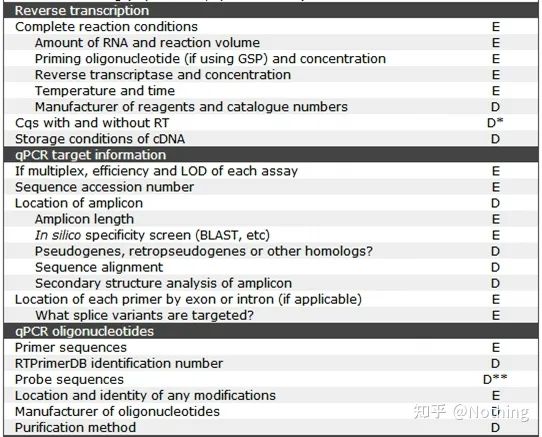
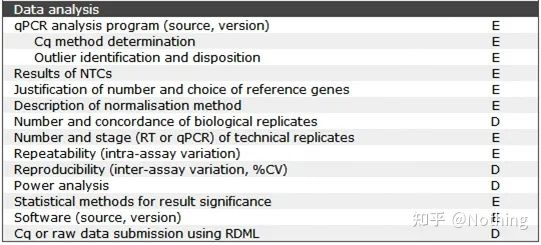
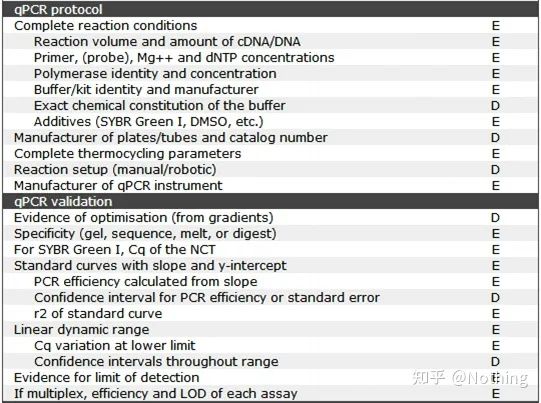
Os revisores podem julgar a qualidade do experimento lendo esses detalhes;futuros leitores também podem usar isso para repetir ou melhorar o experimento.
Vale ressaltar que nesta lista, a importância de cada lista é marcada com E ou D, respectivamente.O que isso significa?E: informações essenciais (devem ser apresentadas);D: informações desejáveis (forneça o máximo possível).
MIQE (1)—Desenho Experimental
Muitos canalhas que concluíram sua defesa após a pós-graduação não saberão como planejar um experimento de forma independente, abrir seus cadernos e fazer o que o professor mandar.Como resultado, o design experimental não foi rigoroso, e o departamento editorial da revista disse que queria fazer essa foto e aquela foto, então o fizeram em transe.É assim que os canalhas são feitos!

Mais perto de casa, o primeiro princípio do experimento é determinaro rigor da lógica experimental.O mais fundamental é o projeto experimental, e o mais importante sobre o projeto experimental é como definir a amostra-alvo, a amostra de referência (controle) e o número de repetições, para que os dados experimentais possam ser referenciados, comparáveis e convincentes.
A amostra alvorefere-se à amostra que nos obriga a detectar o gene alvo após um determinado tratamento.A amostra de referênciaé a amostra sem nenhum tratamento, que muitas vezes é chamada de tipo selvagem na biologia.
Réplicas experimentaissão muito importantes.Geralmente, o número de réplicas persuasivas deve ser superior a três.É necessário distinguir o que é replicação biológica e o que é replicação técnica.
Réplicas biológicas: O mesmo experimento de verificação feito com diferentes materiais (tempo, plantas, lotes, placas de reação).

Duplicação biológica
Tomemos como exemplo o tratamento pesticida da pimenta.Queremos pulverizar pesticidas nas três plantas do ABC, então as três plantas do ABC são três réplicas biológicas, e são o mesmo experimento de verificação realizado com materiais diferentes.Mas como experimento, definitivamente é necessário um controle, então podemos pulverizar um dos ramos da planta A para formar um grupo experimental da planta A, e não pulverizar os outros ramos da planta A para formar um grupo controle.Faça o mesmo para B e C.
Réplicas Técnicas (Réplicas Técnicas): É um experimento repetido projetado para evitar erros causados pela operação, que na verdade é um furo duplicado incluído no mesmo material.Ambos os tratamentos e controles devem ter configurações de replicação (mínimo três) do gene alvo e do gene de referência interno.
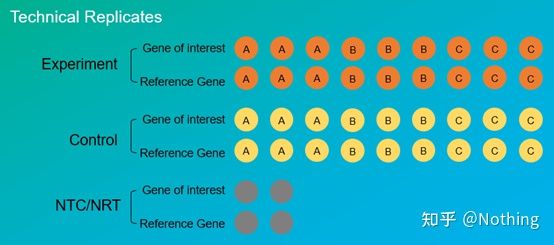
repetição técnica
Pegue a pimenta tratada com pesticidas como exemplo novamente.Para o grupo experimental da planta A, fizemos três furos de PCR de 1, 2 e 3 para seu gene alvo e gene de referência interna, respectivamente, para obter a média após a detecção.Para o controle da planta A, os Grupos também são tratados da mesma forma.Da mesma forma, faça o mesmo tratamento para as plantas B e C.Isso é repetição técnica.
Vale a pena notar queo que entra na estatística é a repetição biológica, e a repetição técnica é para testar se há algum fenômeno aleatório no processo experimental, de forma a tornar os resultados experimentais críveis, ou seja, evitar erros tomando sua média como costumamos dizer.
Controles negativos - NTC e NRT
NTC (controle sem modelo), um controle sem gabarito, é utilizado para verificar se o material experimental está contaminado.Geralmente, a água é usada como modelo.Se houver uma reação fluorescente, isso indica que a contaminação do ácido nucléico ocorreu no laboratório.
Essas poluições vêm de: água impura, reagentes não qualificados contendo DNA endógeno, poluição de primers, poluição de equipamentos de laboratório, poluição de aerossóis, etc., precisam usar removedores de RNase e inibidores de RNase.A poluição por aerossol é a mais difícil de encontrar.Imagine que seu laboratório é como uma névoa, com vários ácidos nucléicos suspensos no ar.

NRT (No-Reverse Transcriptase), o controle sem transcrição reversa, é o RNA transcrito não reverso como um controle negativo, que é o controle do resíduo de gDNA.
Ao fazer a expressão gênica, a quantidade de RNA é detectada pela detecção da quantidade de cDNA após a transcrição reversa.Se houver resíduo de gDNA quando o RNA for purificado, isso causará erros nos resultados experimentais, porque os resultados reais obtidos são gDNA e cDNA.No nível agregado, não apenas cDNA, gDNA precisa ser completamente removido durante a extração de RNA.
MIQE (2) - informações de amostra
As chamadas informações de amostra significam que, quando publicamos um artigo sobre qPCR, devemos explicar claramente as informações de amostra, que são parte indispensável do artigo.Da mesma forma, quando processamos amostras, também devemos regular nossas próprias operações para garantir a validade das amostras.

A descrição da amostra é apenas um resultado, e devemos prestar mais atenção aos materiais retirados durante todo o experimento.
Seleção de materiais experimentais
Amostras de sangue – escolha sangue fresco, não mais que 4 horas.Amostras de células – opte por coletar células frescas em um período de crescimento vigoroso.Tecido Animal—Escolha tecido fresco, de crescimento vigoroso.Tecido Vegetal – Escolha um tecido jovem e fresco.
Você deve ter notado que há uma palavra-chave nessas poucas frases: fresco .
Para as amostras acima, o kit melhor, econômico e estável do mercado é o kit da Foregene, que pode extrair rápida e facilmente seu DNA e RNA.
Kit de isolamento de RNA total da célula
Kit de isolamento de RNA total animal
Kit de isolamento total de RNA vegetal
Kit Plus de Isolamento de RNA Total de Plantas
Kit de isolamento de DNA vegetal
Armazenamento de materiais experimentais
De um modo geral, não recomendamos o armazenamento de amostras, se as condições permitirem.No entanto, existem muitos amigos que não podem realizar experimentos imediatamente após a amostragem e alguns até precisam carregar tanques de nitrogênio líquido para o campo para amostragem.
Para esse tipo de amigo trabalhador, só posso dizer que você não entende de consumíveis de reagentes.Agora, muitas empresas de consumíveis de reagentes produzem reagentes que podem armazenar amostras de RNA à temperatura ambiente e você pode optar por usá-los.O método de armazenamento convencional é o armazenamento de nitrogênio líquido, usando um pequeno tanque de nitrogênio líquido que é fácil de transportar.Depois de trazer a amostra de volta ao laboratório, armazene-a em geladeira a -80°C.

Para experimentos envolvendo RNA, o princípio de seis palavras deve ser seguido:baixa temperatura, sem enzimas,erápido .
O conceito de baixa temperatura é fácil de entender;sem enzimas, a RNase está em todo o mundo em que vivemos (caso contrário, você teria sido morto pelo HIV), então como evitar a RNase ao fazer experimentos é um conceito muito importante;rápido,Não há Kung Fu no mundo que não possa ser quebrado, apenas a velocidade não pode ser quebrada.
Portanto, de certa forma, quanto menor o tempo de extração, melhor o kit.PorqueForegeneO kit enfatiza a velocidade, porque eles a conhecem bem.
PS: Algumas garotas fazem experiências com muito cuidado, mas não são tão boas quanto uma enterrada depois de vários anos de trabalho.Eles sentem que Deus é injusto, reclamando dos outros e procurando a vida.Na verdade, ela não entendeu.Ele não protegeu bem o RNA, e o jogador de slam dunk foi ágil.Quando ele estava fazendo o experimento, ele pensou que terminaria o slam dunk com três vezes, cinco vezes e duas divisões, mas fez bem o experimento.
Observação: Mais lento, mais chances de invasão de RNase.Como treinar para ser rápido?Não tem como, apenas pratique mais.
Para experimentos diferentes e amostras diferentes, ainda é necessário ler mais literatura e escolher um método apropriado para o processamento.Para o processo de coleta e armazenamento de amostras, o MIQE exige que seja claramente escrito no papel, para que os revisores possam avaliar a confiabilidade do papel, e também é conveniente para os jovens atordoados repetirem seu experimento.
Embora os experimentos biológicos sejam difíceis, eles são sofisticados.Se você não for cuidadoso, você pode derrubar o mundo.Por exemplo, transformar a SARS em uma crise bioquímica ou produzir arroz híbrido para salvar 1,3 bilhão de pessoas.A imagem abaixo é um experimento químico, você deve entender o quanto está orgulhoso de sua pesquisa apenas olhando para a aparência de pau dele.Esqueça, não o denegre.

MIQE (3) – extração de ácido nucléico.
A extração de ácido nucleico é um grande evento e todos os experimentos de biologia molecular começam com a extração de ácido nucleico.Em primeiro lugar, vamos copiar o conteúdo do MIQE sobre extração de ácido nucleico.
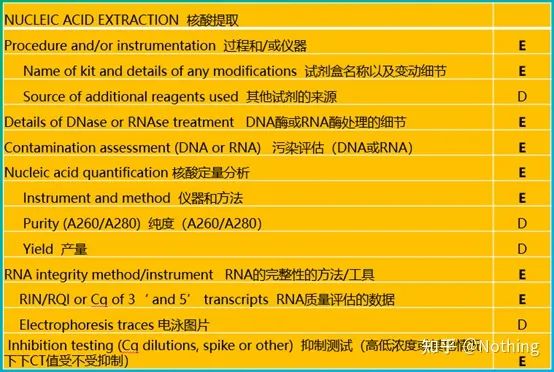
Olhando para esta forma, você não pode ficar na superfície.A forma é um dogma.Para ser um excelente aluno, você deve perguntar por quê.O conteúdo essencial desta tabela é: Perseguira pureza, integridade, consistência e quantidade de extração de RNA .
A primeira parte doprocesso ou instrumento é a etapa de extração de ácido nucleico.Se você usar um extrator automático de ácido nucleico para extrair (avançado, entre em contato comigo para compra), você precisa indicar o nome do modelo do instrumento.

O nome do kit e
qual kit foi usado para os detalhes da mudança, quais reagentes especiais foram adicionados ou quais operações especiais foram feitas devem ser explicados claramente para que outros possam facilmente repetir seu experimento.
Algumas pessoas adicionam alguns reagentes especiais ao extrair amostras especiais, pensando que esta é sua arma secreta e não contam aos outros.Ao mantê-lo em segredo, eles também perdem a oportunidade de fazer seu artigo brilhar.Não seja esperto, você tem que ser mais honesto do que o velho Zhang do campo na pesquisa científica, se você quer ser esperto, o artigo vai te deixar estúpido.
deve lembrar o número do produto do kitquando você encomenda o kit e escreve o artigo.Geralmente, há dois números no kit: Cat—número de catálogo (número do produto, número do artigo), Lot—número do lote do produto (usado para indicar de qual lote o produto veio).
Além disso, o número CAS é frequentemente usado ao solicitar reagentes bioquímicos e vou divulgá-lo juntos.O número CAS é o número dado pela American Chemical Society para cada novo medicamento químico.Geralmente, três números são conectados por um traço.Número CAS de Rushui: 7732-18-5.Os produtos químicos costumam ter vários pseudônimos, mas o número CAS é único.Ao encomendar um medicamento, você pode primeiro verificar seu número CAS.
Mais perto de casa, por que temos que descrever essas coisas claramente?Na verdade, é também para verificar a qualidade da extração de RNA.O uso de instrumentos e kits tornará a extração de RNA mais consistente.A escala de extração dos laboratórios comuns não é grande e pode ser obtida com kits.
Os detalhes do tratamento com DNase ou RNase
A questão importante da PCR quantitativa fluorescente é evitar a contaminação do DNA e não fazer experimentos se houver contaminação.Portanto, é imperativo indicar o processo que você usou para processar o DNA, a fim de demonstrar que o DNA no processo experimental foi completamente removido.representada por um diagrama esquemático.
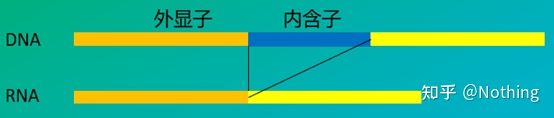
Diagrama esquemático de RNA e DNA
Em geral, o método para remover o DNA é tratar o RNA com DNase após a extração.No entanto, estes são métodos relativamente antigos.Os kits comerciais de extração de RNA conseguiram remover o DNA durante o processo de extração sem adicionar DNase.Por exemplo, uma série de kits da Foregene .
Observação: A remoção do DNA durante a extração do RNA é uma faca de dois gumes muito perigosa, que prolongará o tempo de operação da extração do RNA e aumentará o risco de degradação do RNA.Basicamente, é uma compensação entre o rendimento e a pureza do RNA.
Além disso, a quantidade de DNase adicionada à coluna de adsorção à base de sílica é muito pequena e DNase de alta qualidade deve ser usada para obter o efeito.A DNase não otimizada não pode ser digerida rápida e completamente.Este é um teste de nível técnico do comerciante.Claro, existem comerciantes ainda mais estranhos que se gabam de que o DNA pode ser removido sem DNase.Pode-se dizer que quem se gaba de que o DNA pode ser completamente removido sem a DNase é um hooligan.O DNA é uma estrutura de fita dupla relativamente estável e não pode ser eliminada apenas com conversas e risadas.
Avaliação de contaminação
método de avaliação: detecção por eletroforese, 1% de agarose, 6V/cm, 15min, carregamento de 1-3 ul
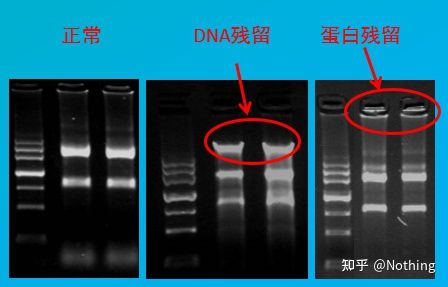
Análise quantitativa de ácido nucleico
geralmente é medido usando um espectrofotômetro UV.Deixe-me primeiro popularizar o significado dos três valores de OD260, OD280 e OD230.
·OD260nm: É o comprimento de onda de absorção do pico de absorção mais alto do ácido nucléico, e o melhor valor medido varia de 0,1 a 1,0.Caso contrário, dilua ou concentre a amostra para trazê-la dentro da faixa.
·OD280nm: É o comprimento de onda de absorção do maior pico de absorção de proteínas e substâncias fenólicas.
·OD230nm: É o comprimento de onda de absorção do maior pico de absorção de carboidratos.
A seguir, vamos falar sobre o papel de cada indicador.Para A260, pode ser usado para medir o rendimento do ácido nucleico.Quando OD260=1, dsDNA=50μg/ml, ssDNA=37μg/ml, RNA=40μg/ml.
Para pureza, precisamos observar as proporções que normalmente vemos: OD260/280 e OD260/230.
·DNA puro: OD260/280 é aproximadamente igual a 1,8.Quando for maior que 1,9, indica que há poluição de RNA, e quando for menor que 1,6, indica que há poluição de proteínas e fenóis.
·ARN puro: 1,7
·OD260/230: Seja DNA ou RNA, o valor de referência é 2,5.Quando for menor que 2,0, indica que há poluição por açúcar, sal e matéria orgânica.
Integridade do RNA
É muito importante medir a integridade do RNA.Geralmente, é necessário fazer um experimento de gel de desnaturação de RNA para verificar se o brilho entre 28S e 18S RNA é uma relação dupla.Quando a terceira banda 5S aparece, significa que o RNA começou a se degradar, exceto para invertebrados.

Dados para avaliação da qualidade do RNA: Além dos testes acima, existem também alguns testes de instrumentos mais avançados em termos de integridade do RNA, como o teste de integridade RQI do sistema de eletroforese automática Experion, que pode detectar se o RNA é degradado de forma invisível.
Na pesquisa científica, PCR quantitativo fluorescente é uma comparação entre o gene alvo e o gene de referência interno.Portanto, no processo de preservação de amostras de RNA, extração de RNA, etc., o objetivo principal é garantir a integridade do RNA.
Como a integridade do RNA afeta o equilíbrio entre o gene alvo e o gene de referência interna pode ser facilmente entendido na figura abaixo.A degradação levará à incompletude do gene, seja a incompletude do gene de referência interno ou a incompletude do gene alvo, terá um grande impacto nos dados.

O diagrama esquemático do gene alvo e do gene de referência não deve ser verdadeiro
Teste de inibição (se o valor CT é suprimido sob alta ou baixa concentração ou outras condições)
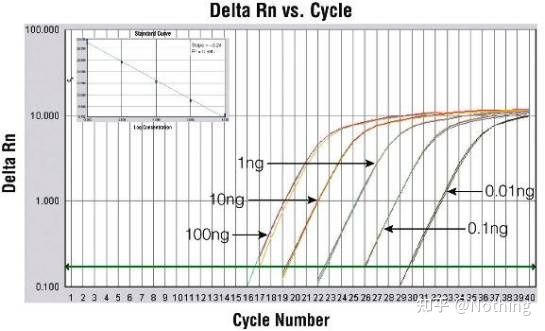
Tomando esta figura como exemplo, os valores de Ct das cinco curvas são os seguintes.A distribuição dos valores de CT entre as curvas é desigual, e os valores de Ct são retardados em altas e baixas concentrações, que é o caso da inibição da PCR.
Ponto-chave: No processo de extração de RNA, precisamos abandonar os equívocos e estabelecer os corretos.
A ideia errada é: a extração de RNA só persegue o rendimento, pensando que quanto maior a quantidade de RNA obtida, melhor.Na verdade, quando fazemos quantificação, se o número de genes não for muito grande, não precisamos de muito RNA.A quantidade de RNA que você extrai é mais do que suficiente.
O conceito correto é:A extração de RNA deve buscar pureza, integridade e consistência.A pureza pode garantir que a transcrição reversa subsequente não seja inibida e os dados não sejam afetados pelo DNA.A integridade garante o equilíbrio das sequências-alvo e das referências internas.A consistência garante carregamento de amostra estável.
MIQE (4) – transcrição reversa
Equívoco: a busca de maior volume de amostra.
conceito correto: Busque a consistência (estabilidade), independentemente da quantidade de RNA carregado, a eficiência da transcrição reversa permanece consistente, garantindo que as diferenças no cDNA possam realmente refletir as diferenças no mRNA.
Explicamos esse processo com um diagrama esquemático:
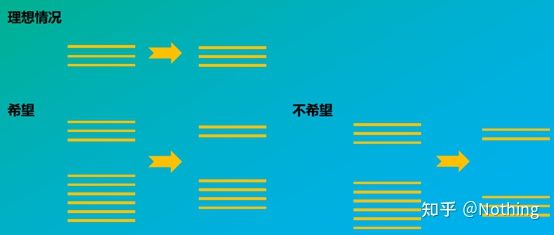
Diagrama esquemático da eficiência da transcrição reversa, não é verdade
Em primeiro lugar, precisamos entender a diferença entre o processo de transcrição reversa e o processo de PCR.A PCR passa por vários processos de aquecimento e recozimento, e o fragmento alvo cresce exponencialmente;embora a transcrição reversa não tenha esse processo, podemos imaginar que a transcrição reversa é na verdade um-para-um Durante o processo de replicação, tantos pedaços de RNA
como existem tantos fragmentos de informações de cDNA, isso deve ser entendido agora, porque fragmentos grandes e pequenos foram transcritos reversamente e é impossível focar em um fragmento.E como a quantidade de RNA é relativamente pequena, a quantidade de cDNA obtida também é relativamente pequena, ao contrário do PCR, que tem um efeito de amplificação, portanto é basicamente impossível de detectar.
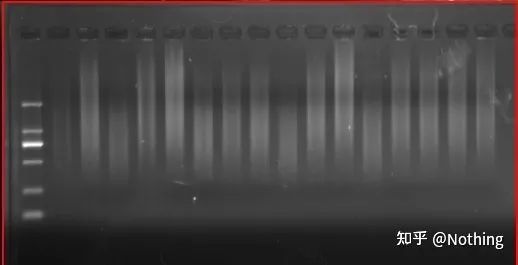
resultados da eletroforese de cDNA
Em segundo lugar, idealmente, a transcrição reversa é realizada individualmente, mas nenhuma transcriptase reversa de qualquer empresa pode alcançar esse efeito.Basicamente, a eficiência da maioria das transcriptases reversas oscila entre 30-50%.Se for esse o caso, preferimos ter uma eficiência de transcrição reversa relativamente estável, que é o que queremos ver na figura: 3 RNAs obtêm 2 cDNAs, 6 RNAs obtêm 4 cDNAs, portanto, não importa quanta amostra seja carregada, a eficiência da transcrição reversa é relativamente estável.Não queremos ver a situação em que a eficiência da transcrição reversa é instável e a alta concentração é inibida.
Então, como verificar se a eficiência da transcrição reversa é estável?O método é muito simples, você só precisa fazer um teste de comparação: um é fazer a transcrição reversa em cDNA após duplicar a diluição do RNA, e o outro é fazer a diluição dupla após a transcrição reversa em cDNA e depois fazer qPCR para ver a inclinação obtida é consistente.Como um excelente aluno, você deve entender isso em segundos.Como mostrado abaixo:
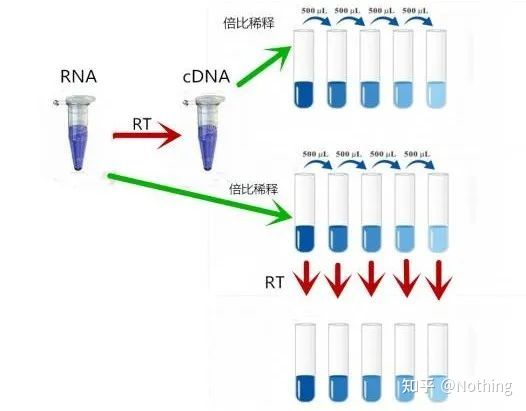
Diluição de RNA e cDNA para testar se a eficiência da transcrição reversa é estável
Transcriptase reversa e kit
Como o PCR quantitativo fluorescente perfeito pode ter transcriptase reversa e kit excelentes.A transcriptase reversa é dividida em dois tipos, de acordo com a fonte, AMV ouM-MLV, e seu desempenho é o mesmo mostrado na tabela.
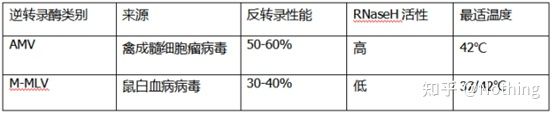
Atividade de RNase H
RNase H é Ribonuclease H, o nome chinês é ribonuclease H, que é uma endoribonuclease que pode hidrolisar especificamente o RNA na cadeia híbrida DNA-RNA.A RNase H não pode hidrolisar as ligações fosfodiéster em DNA ou RNA de fita simples ou dupla, ou seja, ela não pode digerir DNA ou RNA de fita simples ou dupla.Comumente usado na síntese da segunda fita de cDNA.
É uma coisa estranha.Dizemos que a transcriptase reversa tem atividade de RNase H, não que a transcriptase reversa contenha RNase H, e pode não ser possível separar a RNase H da transcriptase reversa, talvez devido à conformação de certos grupos na transcriptase reversa.
Portanto, independentemente da maior eficiência de transcrição reversa do AMV, sua atividade de RNase H reduz o rendimento do cDNA.Obviamente, os fabricantes de reagentes estão constantemente otimizando seus produtos para eliminar a atividade da RNase H na transcriptase reversa tanto quanto possível para aumentar o rendimento do cDNA.
temperatura de recozimento
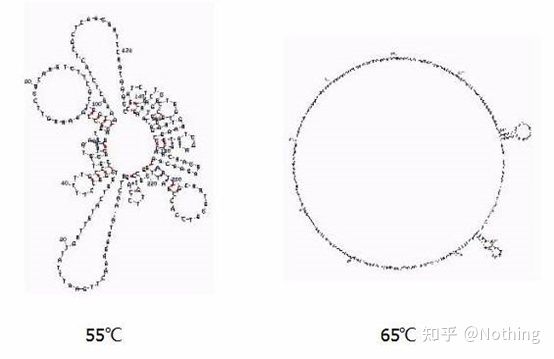
Estrutura secundária do RNA em diferentes temperaturas
Veja a figura acima para a estrutura secundária do RNA em diferentes temperaturas e use a ferramenta on-line mFold para determinar a estrutura secundária do fragmento alvo sob condições específicas de temperatura e concentração de sal.A 55°C, a estrutura secundária do RNA ainda é muito complexa, a transcriptase reversa não pode funcionar e a estrutura secundária não pode ser completamente resolvida até 65°C, enquanto a temperatura ótima de AMV e M-MLV é muito menor do que esta temperatura.
o que fazer?A estrutura secundária é o pareamento complementar do próprio template, o que leva a uma forte competição entre o primer e a transcriptase reversa e o template, resultando em uma série de problemas como baixo E e baixa repetibilidade.
o que fazer?Aumente apenas a temperatura de recozimento tanto quanto possível.
Muitos fabricantes de reagentes estão melhorando sua transcriptase reversa por meio da engenharia genética.Alguns aumentam a temperatura da reação, como Jifan e Aidelai, e alguns removem o grupo ativo da enzima RNase H para melhorar a afinidade entre a enzima e o molde de RNA.A alta afinidade pode espremer competitivamente a estrutura secundária e ler sem problemas, além de melhorar muito a eficiência da transcrição reversa.
Ponto-chave: A transcrição reversa é mais importante para buscar a consistência da eficiência da transcrição reversa (as enzimas não devem apenas ser eficientes, mas também estáveis), em vez da quantidade de amostra carregada, se não for uma PCR quantitativa fluorescente particularmente em grande escala, não será possível.Múltiplos cDNAs.
Vários fabricantes também fizeram alguns esforços na busca de consistência.Por exemplo, a maioria das empresas agora embala a transcrição reversa como um kit padrão para venda, o que é uma boa escolha.
Por exemplo, os kits RT Easy Series da Foregene:
RT Easy I (pré-mistura principal para kit de síntese de cDNA de primeira fita)
MIQE (5) - informações do gene alvo

A figura acima explica
1. Se este gene é eficaz para experimentos repetidos geralmente pode ser verificado por experimentos repetidos.
2. ID do gene, você sabe.
3. Comprimento do gene, o comprimento total do gene alvo definitivamente não é problema.Ao projetar primers, certifique-se de que o comprimento do amplicon esteja entre 80-200bp para garantir uma melhor eficiência de amplificação.
4. Informações de comparação do Sequence Blast, o gene alvo precisa ser comparado no banco de genes para evitar amplificação não específica.
5. Presença de pseudogenes.Um pseudogene é uma sequência de DNA semelhante a um gene normal, mas perde sua função normal.Frequentemente existe na família multigênica dos eucariotos.Geralmente é representado por ψ.É uma cópia de DNA genômico não funcional no genoma que é muito semelhante à sequência do gene codificante., geralmente não são transcritos e não têm significado fisiológico claro.
6. Posição dos primers em relação aos exons e introns.Nos primeiros anos, quando resolvíamos o problema da contaminação do DNA, muitas vezes prestávamos atenção às posições dos primers, éxons e íntrons e geralmente considerávamos projetar primers nos íntrons para evitar a amplificação do DNA.Por favor, veja a figura abaixo: preto representa íntrons, vários azuis representam éxons, rosa representa primers comuns e vermelho brilhante representa primers abrangendo íntrons.
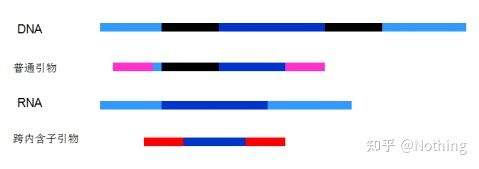
Esquema, nunca verdadeiro
Que plano perfeito isso parece, mas na verdade, na maioria dos casos, os primers trans-intron não são tão mágicos quanto se imagina, e eles também causarão amplificação não específica.Portanto, a melhor maneira de prevenir a contaminação do DNA é removê-lo completamente.
7. Previsão de conformação.Usando este exemplo novamente, use a ferramenta online mFold para determinar a estrutura secundária do fragmento alvo em uma temperatura e concentração de sal específicas.
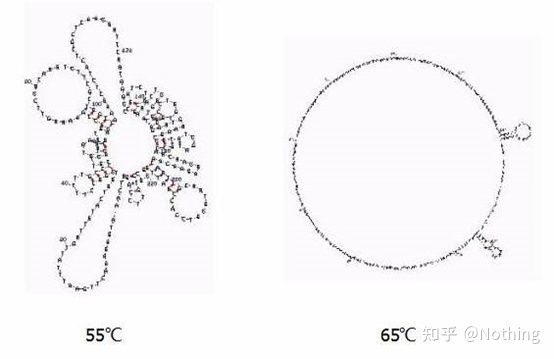
Estrutura secundária do RNA em diferentes temperaturas
A estrutura secundária é o pareamento complementar do próprio template, o que levará a uma forte competição entre o primer e o pareamento do template, e as chances de ligação do primer são menores, resultando em uma série de problemas como baixo E e baixa repetibilidade.Através da previsão de software, se não houver nenhum problema de estrutura secundária, isso seria ótimo.Se houver, nosso artigo de acompanhamento discutirá especificamente como resolver esse problema.
MIQE (6)—qPCR Oligonucleótidos

Para PCR quantitativo fluorescente, a primeira coisa com a qual você luta todos os dias é a extração de RNA, e a segunda coisa pode ser o design do primer.
Em primeiro lugar, ainda verificamos as regras sobre o design do primer de acordo com a lista de verificação do MIQE.É tão simples que os canalhas podem rir, e podemos terminar em uma frase: descubra a sequência e posição da sonda primer e o método de modificação.Para o método de purificação de primer, a síntese de primer é tão barata no momento, qPCR é digna de PAGE e métodos de purificação acima, e as informações do instrumento de síntese não são importantes.Muitas pessoas fazem primers há décadas e não sabem que o sintetizador é o ABI3900.
Em relação aos princípios do design de primers, você não precisa memorizá-los automaticamente, porque a maioria dos softwares de design de primers ou ferramentas on-line podem cuidar desses problemas (ferramenta on-line recomendada primer3.ut.ee/) e 99,999% do design de primers não é feito manualmente.
Basta verificar os seguintes pontos após a concepção dos primers:
1. Desenhar primers perto da extremidade 3': No caso de usar primers oligo dT para a síntese da primeira cadeia de cDNA, considerando a eficiência da transcrição reversa e a integridade do RNA, os primers projetados precisam ser projetados perto da extremidade 3' para melhorar a eficiência da amplificação.Use uma imagem para explicar da seguinte forma (não há como entender isso):
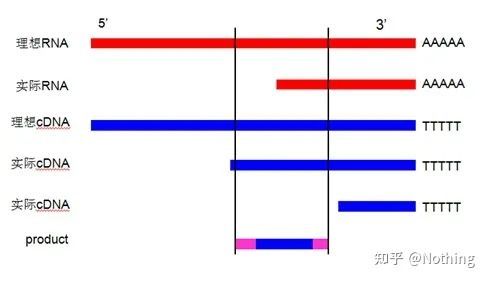
Por que os primers devem ser projetados perto da extremidade 3', isso não deve ser verdade
2. Valor TM: O valor Tm está em 55-65°C (porque a atividade da exonuclease é mais alta em 60°C), e o conteúdo de GC está em 40%-60%.
3. BLAST: Para evitar a amplificação não específica do genoma, o Blast deve ser usado para verificação complementar.
MIQE(7)—processo qPCR
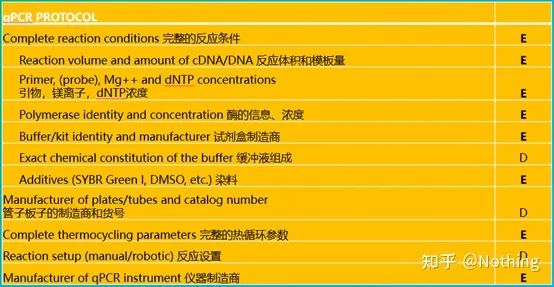
1. kit qPCR
De acordo com os requisitos do MIQE, devemos descrever claramente as condições completas da reação no artigo, incluindo a configuração do sistema de reação de PCR, qual kit é usado, quem é o fabricante, qual é o tamanho do sistema de reação, se o método de corante ou o método de sonda é usado, configurações do programa de PCR.Os motoristas veteranos definitivamente descobrirão que, desde que o kit seja selecionado, as informações acima são basicamente determinadas.
Atualmente, a fabricação e produção de kits PCR quantitativos fluorescentes é uma tecnologia muito madura.Desde que você não escolha fabricantes extremamente ruins, a probabilidade de problemas não é alta, mas ainda queremos compartilhar com você alguns pontos:
Enzima Taq de arranque a quente:A parte mais importante da PCR é a enzima Taq hot-start.As enzimas hot-start no mercado são geralmente divididas em dois tipos, uma é uma enzima hot-start quimicamente modificada (você pode imaginá-la como embebida em parafina) e a outra é uma enzima hot-start para modificação de anticorpos (ligação antígeno-anticorpo).A modificação química é uma forma inicial de enzimas de partida a quente.Quando uma certa temperatura é atingida, a enzima libera sua atividade.A enzima de arranque a quente modificada por anticorpo usa métodos biológicos para bloquear a atividade da enzima.Quando uma certa temperatura é atingida, o anticorpo será desnaturado e inativado como uma proteína, e a atividade enzimática entrará em ação.
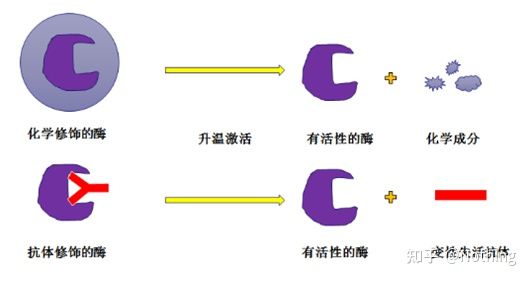
No entanto, qual é a utilidade disso?Este é o caso, a atividade de liberação de enzimas modificadas por anticorpos é mais rápida do que as enzimas modificadas quimicamente, portanto, em termos de sensibilidade, as enzimas modificadas por anticorpos têm uma pequena vantagem, de modo que basicamente não há enzimas modificadas quimicamente nos kits no mercado.Se houver, a tecnologia deste fabricante ainda está presa na era do milênio.
Concentração de íons de magnésio:A concentração de íons de magnésio é muito importante na reação de PCR.A concentração adequada de íons de magnésio pode promover a liberação da atividade da enzima Taq.Se a concentração for muito baixa, a atividade da enzima será significativamente reduzida;se a concentração for muito alta, a amplificação não específica catalisada por enzima será aumentada.A concentração de íons de magnésio também afetará o recozimento dos primers, a temperatura de fusão do molde e os produtos de PCR, afetando assim o rendimento dos fragmentos amplificados.A concentração de íons de magnésio é geralmente controlada em 25mM.Obviamente, para um bom kit, a concentração de íons de magnésio deve ser bem controlada.Alguns comerciantes adicionam um agente quelante de íons de magnésio ao reagente, o que pode alcançar o efeito de ajuste automático da concentração de íons de magnésio.
Concentração de corante fluorescente:O corante fluorescente, que é o SYBR Green que costumamos usar, gera fluorescência principalmente pela ligação ao sulco menor do DNA de fita dupla, porque a ligação do corante ao DNA de fita dupla é inespecífica, ou seja, desde que o DNA de fita dupla seja combinado com ele, a fluorescência pode ocorrer, então primer-dímeros e modelos de DNA no sistema se combinarão com ele para formar um sinal de fundo.
PS: Devido às suas propriedades fotossensíveis, os produtos no mercado geralmente são embalados em tubos de centrífuga opacos de cor marrom (conforme a figura abaixo).No entanto, isso encontrará um problema.É difícil ver se o líquido é sugado durante a amostragem.A este respeito, Qingke é de fato o mais fácil de usar (como mostrado na figura abaixo), e o tubo transparente é embalado em um saco de estanho opaco.Em seguida, coloque-o em um saco de lata, levando em consideração a conveniência de evitar luz e amostragem.Você deve escolher o número de produto correto.TSE204 é uma existência super econômica, o que me faz querer plantar grama.

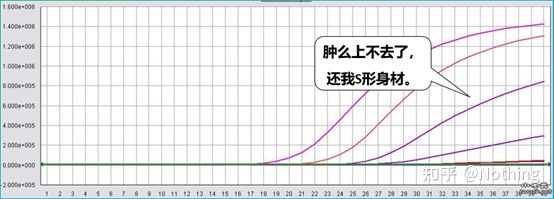
A concentração do corante fluorescente também é muito importante.Se a concentração for muito baixa, a curva de amplificação não subirá no estágio posterior e não será perfeita;se a concentração for muito alta, causará interferência de ruído.Uma vez que a PCR quantitativa fluorescente depende principalmente do valor CT, se a concentração do corante fluorescente não for ajustada adequadamente, o ponto baixo é melhor que o ponto alto.Obviamente, a concentração de corante apropriada é a melhor.
ROX: os corantes ROX são usados para corrigir erros de sinal de fluorescência entre poços.Alguns fabricantes de instrumentos exigem calibração, enquanto outros não.Por exemplo, o uso do instrumento de amplificação de PCR em tempo real da Thermo Fisher Scientific geralmente requer calibração, incluindo 7300, 7500, 7500Fast, StepOnePlus, etc. As instruções gerais do kit irão descrevê-lo.
O qPCR Mix da Foregene também contém corante ROX, que é conveniente para uso em vários modelos.
Kit de PCR em tempo real-Taqman
Tratamento de ligação de hidrogênio fraca: O tratamento de ligações de hidrogênio fracas é um assunto relativamente técnico.Nada leu os manuais de muitos kits, mas nenhum deles mencionou este tópico.Na verdade, é tão importante.A combinação de bases depende principalmente da força das ligações de hidrogênio.Pontes de hidrogênio fortes são amplificações normais, e ligações de hidrogênio fracas levam a amplificações não específicas.Se as ligações de hidrogênio fracas não puderem ser bem eliminadas, a amplificação não específica não poderá ser evitada.No escopo do autor, apenas algumas empresas perceberam esse problema.Ao comprar o kit, você pode consultar se considerou uma solução nesse sentido para o kit que deseja escolher.
volume de reação: O sistema de 20-50ul é mais comumente usado e volumes menores provavelmente causarão erros.De um modo geral, as instruções do kit recomendam o uso de volumes de reação de PCR.Não seja esperto e use volumes menores para economizar custos.O objetivo de.O volume recomendado pelos comerciantes foi realmente testado, e pode ser que eles não consigam resolver o problema de erros causados por pequenos volumes.
2. O fabricante e o número do artigo da placa do tubo
Todos conhecem o princípio da PCR quantitativa fluorescente.A coleta de fluorescência é realizada principalmente por meio de tampas de tubos de PCR.Ao escolher consumíveis de PCR, preste atenção a dois pontos: boa transmissão de luz e adequado para o instrumento.De um modo geral, as pranchas e tubos das marcas tradicionais são boas, mas você tem que escolher com cuidado em termos de adaptação, caso contrário, não conseguirá usar o instrumento.

4. Conhecimento de alto nível
MIQE (8) - validação qPCR
Esta é a principal prioridade do qPCR!Tantos heróis caíram na areia aqui.Claro, também é possível que você tenha sorte e os genes que você estudou sejam simples, então você flutuou pela caverna de gelo ao longo do vento.As informações de verificação do qPCR destinam-se a testar a confiabilidade dos dados.Listamos as informações de verificação necessárias da seguinte forma:
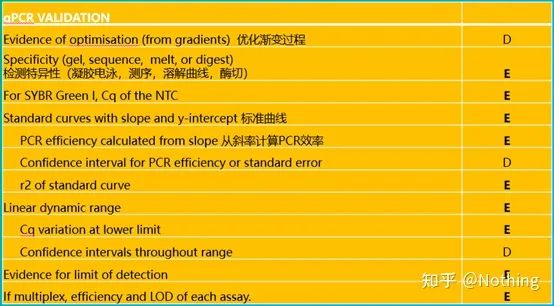
1. Teste de especificidade
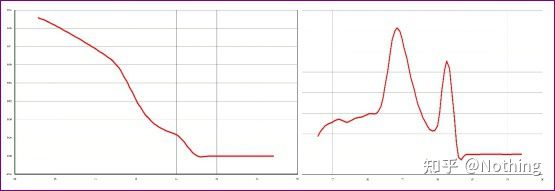
A especificidade da amplificação do gene alvo é testada verificando se a imagem da eletroforese é uma única banda;verificação de sequenciamento;curva de fusão para ver se o mapa de pico é único;verificação de digestão enzimática e outros métodos.
Aqui, nos concentramos em tanálise da amplificação inespecífica pelo método das curvas de fusão.De um modo geral, quando projetamos primers, o tamanho do fragmento do produto deve estar na faixa de 80-200 pb, o que torna a temperatura de fusão do produto de PCR na faixa de 80-85 °C.Portanto, se houver picos diversos, deve haver outros produtos de amplificação não específicos;se o pico aparecer abaixo de 80°C, geralmente é considerado um dímero de primer;se o pico aparecer acima de 85°C, geralmente é considerado contaminação de DNA ou mais amplificação inespecífica de grandes fragmentos.
Nota: Às vezes, há apenas um único pico a 80°C.Neste momento, este conceito deve ser respeitado.É provável que os resultados da amplificação sejam todos dímeros de iniciadores.
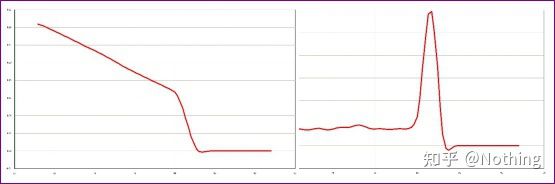
Curva de fusão normal (pico único sem amplificação não específica)
Curva de fusão problemática (amplificação não específica de picos espúrios)
【Análise de caso】
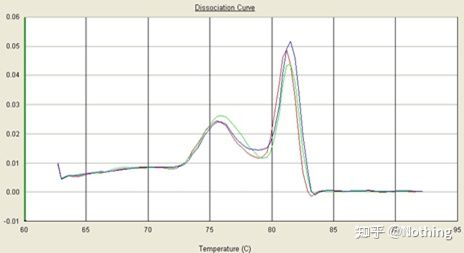
Há um pico principal, mas o dímero do primer é sério
A curva de fusão de pico único na figura abaixo pode facilmente enganar seus olhos, pensando que é um experimento perfeito, mas o resultado está completamente errado.Neste momento, temos que olhar para a temperatura de fusão.A temperatura máxima é inferior a 80°C, que é completamente primer-dímero.
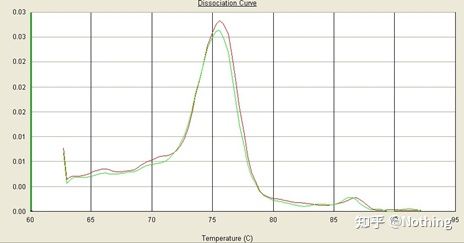
Nenhum fragmento alvo, todos os dímeros de primer
Aqui, meu irmão não pode parar.A foto abaixo é uma foto tirada com um celular que me foi enviada por um canalha.Os reagentes que ele usou são todos de marcas comumente usadas na indústria.Ele mudou de uma marca de prefixo T para outra marca de prefixo T.Acho que você já adivinhou.O canalha gritou para mim: “O reagente usado na primeira foto é bom demais e o pico é único.Mais tarde, depois de usar o reagente que você recomendou, fica como na segunda foto, com picos mistos.Você me deixou infeliz.“
Separe os dois gráficos.À primeira vista, um tem um pico único e o outro tem um pico duplo.Bobagem, um único pico é claro.Isso é verdade?
Pior que Dou E, se eu colocar as duas fotos na foto abaixo, vocês vão entender na hora.Na verdade, ficamos facilmente paralisados por esse tipo de imagem.Após uma análise cuidadosa, descobrimos que: o pico da primeira figura está em 75°C, que é completamente dímero de primer;o pico da segunda figura aparece em 75°C e 82°C, pelo menos há O produto aparece.
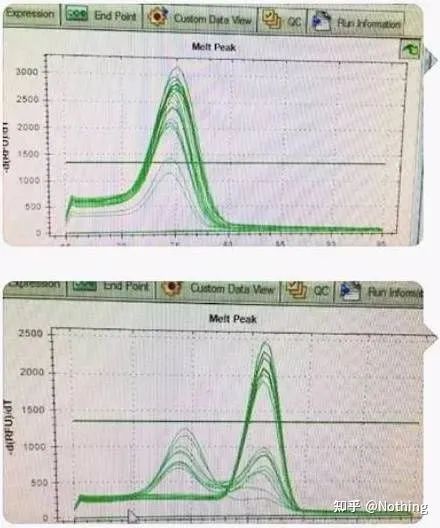
Fotos do feedback dos alunos
Portanto, o problema fundamental não é o problema dos reagentes, mas o problema do design do primer.Ao mesmo tempo, também prova que algumas grandes marcas não são de qualidade de ferro, e também prova o que meu irmão disse antes: Não é a marca de reagente que apóia seu artigo.É o seu artigo que sustentou a marca dos reagentes.Imagine só, se o canalha não trocasse os reagentes, os dados errados seriam enviados para o diário, e o que aconteceria seria uma tragédia.
2. Valor Ct do controle em branco
Não explique, se o controle em branco tiver um valor Ct, isso não é poluição?No entanto, você ainda precisa entender qual controle em branco tem um valor Ct.Se for NTC, significa que há DNA estranho, como contaminação de reagente.Se for NRT, significa que o RNA extraído tem contaminação de DNA.
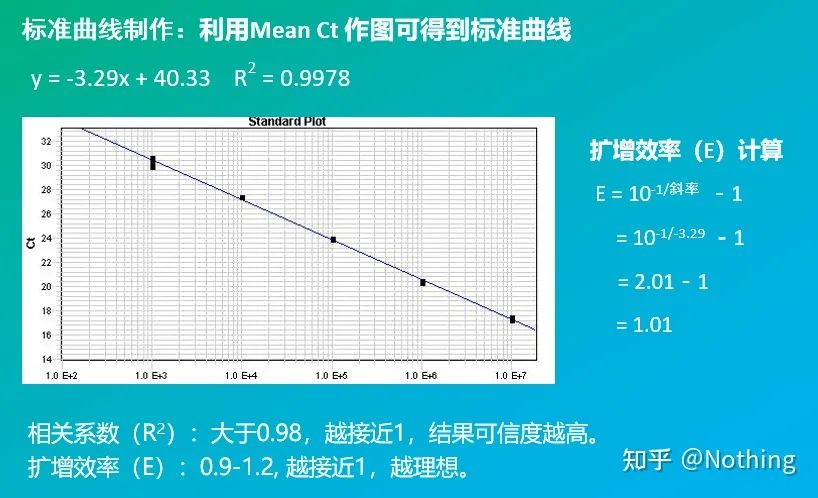
3. Curva padrão
Incluindo a inclinação e a fórmula de cálculo, a eficiência do PCR pode ser calculada por meio da fórmula.Um experimento perfeito requer que a inclinação da curva padrão se aproxime de 3,32 e R² se aproxime de 0,9999.
4. Faixa dinâmica linear
A faixa dinâmica da reação é linear.De acordo com o modelo usado para gerar a curva padrão, a faixa dinâmica deve incluir pelo menos 5 gradientes de concentração e prestar atenção à alteração dos valores de Ct em gradientes de alta concentração e gradientes de baixa concentração.
5. Precisão de detecção
As alterações nos resultados do qPCR, ou seja, baixa repetibilidade, ou seja, baixa precisão, são causadas por muitos fatores, incluindo temperatura, concentração e operação.A precisão do qPCR geralmente se torna menos controlável à medida que o número de cópias diminui.Idealmente dentro da variação experimental, essa variação técnica deve ser distinta da variação biológica, e as réplicas biológicas podem abordar diretamente as diferenças estatísticas nos resultados de qPCR entre grupos ou tratamentos.Em particular para ensaios de diagnóstico, a melhor precisão entre ensaios (repetibilidade) entre locais e operadores deve ser relatada.
6. Eficiência de detecção e LOD (em multiplex qPCR)
LOD é a menor concentração de 95% das amostras positivas detectadas.Em outras palavras, a concentração de LOD contida em um conjunto de replicações do gene alvo não deve exceder 5% das reações que falharam.Ao fazer análise multiplex qPCR, especialmente para detecção simultânea de mutações pontuais ou polimorfismos, multiplex qPCR precisa fornecer evidências de que a precisão de vários fragmentos de alvo não é comprometida no mesmo tubo, detecção múltipla e detecção de tubo único. Eficiência e LOD devem ser os mesmos.Especialmente quando genes-alvo de alta concentração e genes-alvo de baixa concentração são amplificados simultaneamente, esse problema deve ser levado em consideração.
Problemas e soluçõesDe um modo geral, os problemas frequentemente encontrados na depuração qPCR se concentram nos seguintes aspectos:
· amplificação inespecífica
· Dificuldade na escolha da concentração de primer e problemas com dímeros de primer
·A temperatura de recozimento é imprecisa
·Estrutura secundária afeta a eficiência de amplificação
amplificação inespecífica
amplificação não específicaocorre , geralmente é considerado se o design do primer não é adequado, mas se você não estiver com pressa para trocar os primers, tente primeiro os seguintes métodos (o princípio também está anexado):
·Aumentar a temperatura de recozimento – tentar fazer ligações de hidrogênio fracas incapazes de manter;
·Encurte o tempo de recozimento e alongamento - reduza a chance de ligações de hidrogênio fracas;
·Reduz a concentração de primers – reduz a chance de ligação de primers redundantes e regiões não-alvo;
Baixa eficiência de amplificação
A situação oposta à amplificação não específica – baixa eficiência de amplificação , e as medidas para lidar com baixa eficiência de amplificação são exatamente o oposto:
·Prolongar o tempo de recozimento e alongamento;
·Altere para PCR de três etapas e reduza a temperatura de anelamento;
·Aumentar a concentração do primer;
Ps: Muitos estudantes de pós-graduação nascidos na década de 90 não estão dispostos a estudar como depurar experimentos e esperam que o kit resolva completamente o problema (se você quiser ir a uma empresa de reagentes para fazer pesquisa e desenvolvimento após a formatura);Para resolver facilmente o problema, os tolos ainda precisam ler a introdução da empresa de reagentes para ver se existe um fator que absorve as ligações de hidrogênio fracas.
Escolha difícil de concentração de primer e problemas com dímeros de primer
Método 1: De um modo geral, as instruções do kit para qPCR têm sistemas recomendados e concentrações recomendadas de primers.
Método 2: Depuração definindo o gradiente de concentração do primer.A foto abaixo foi roubada de uma empresa para ilustrar.A figura abaixo mostra os resultados quantitativos de fluorescência feitos com três gradientes de concentração de primer (100 nM, 250 nM, 500 nM) e quatro gradientes de concentração de modelo (0,1 ng, 1 ng, 10 ng, 100 ng).O valor Ct dos resultados experimentais é plotado da seguinte forma:
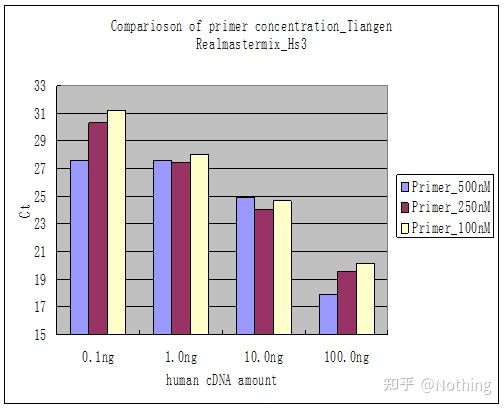
Seleção de concentração de primer Concatene cada concentração de primer em uma linha como segue:
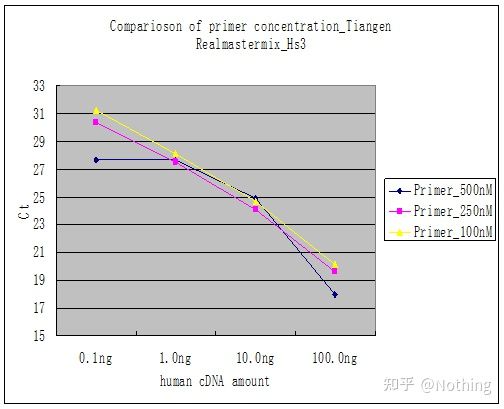
A escolha da concentração do primer é óbvia, a relação linear da concentração do primer de 100 nM e 250 nM é melhor e a relação linear da concentração do primer de 500 nM é relativamente ruim.Em 100 nM e 250 nM, o valor Ct de 250 nM é relativamente pequeno, então a concentração ideal de primer é 250 nM.Em geral, dímeros de primer severos podem ser vistos na curva de fusão.E se os primers projetados não puderem evitar os dímeros de primer?
Método 3: Reduza a quantidade de primers e aumente a temperatura de recozimento (não precisa explicar).
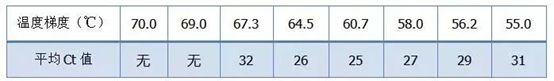
O valor empírico da temperatura de recozimento é 60°C.Se você não tiver certeza, como selecionar uma temperatura de recozimento mais adequada?A resposta é a mesma que a escolha da concentração do primer -teste de gradiente.Tire uma foto da empresa Bio-rad para ilustrar o problema.Para a amplificação de um determinado fragmento alvo, defina oito gradientes de temperatura, cada um com três repetições, e a curva de amplificação obtida é a seguinte:
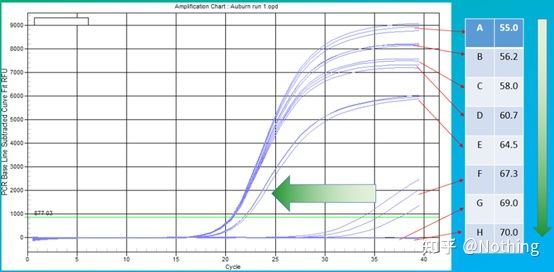
seleção de temperatura de recozimento:
·70°C, 69°C—Basicamente, os primers não podem ser combinados, então não há amplificação.
·67,3°C – Há uma pequena quantidade de amplificação no início, e o valor de Ct é relativamente grande.
·64,5°C——O valor Ct diminui.
·A 60,7°C, 58,0°C, 56,2°C e 55,0°C, os valores de Ct basicamente tenderam a ser estáveis, mas os valores finais de fluorescência foram diferentes.
Como escolher?Princípio: O primeiro princípio é o valor Ct mais alto.Para o mesmo valor de Ct, escolha uma temperatura de recozimento mais alta para evitar dimerização e amplificação não específica.Embora haja um valor de fluorescência maior a 55°C, pode haver dímeros ou amplificação inespecífica na mesma.
Mas se você for tão esperto quanto você, com certeza vai pensar: Logicamente falando, se a reação de PCR for muito específica, desde que a concentração do primer exceda o requisito mínimo, os pontos altos e baixos não devem ter efeito, assim como corantes fluorescentes e dNTPs.De fato, desde que a temperatura de recozimento seja otimizada adequadamente, o efeito da concentração do primer no valor de Ct será naturalmente minimizado.
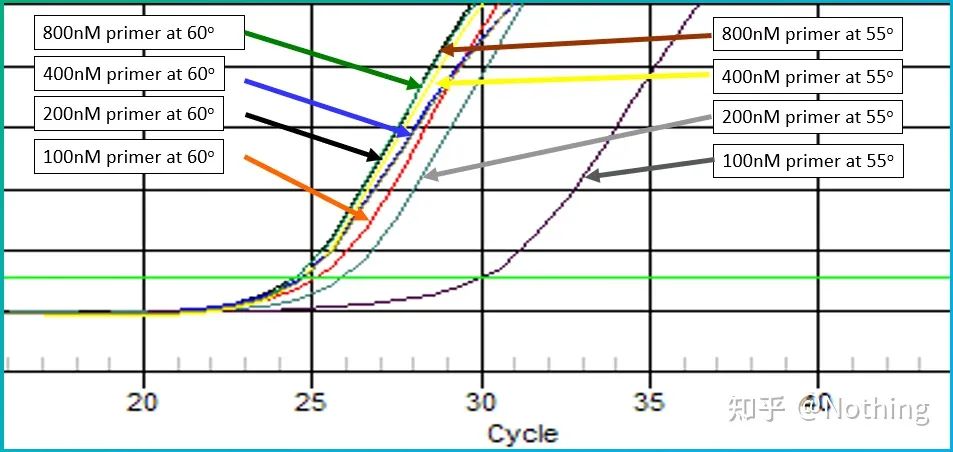
A temperatura de recozimento é otimizada adequadamente e o efeito da concentração do primer no CT será minimizado
A estrutura secundária afeta a eficiência da amplificação
Vamos pegar a foto da Bio-rad para ilustrar o problema.Ele também projeta um gradiente de temperatura para amplificar um gene com uma estrutura secundária.
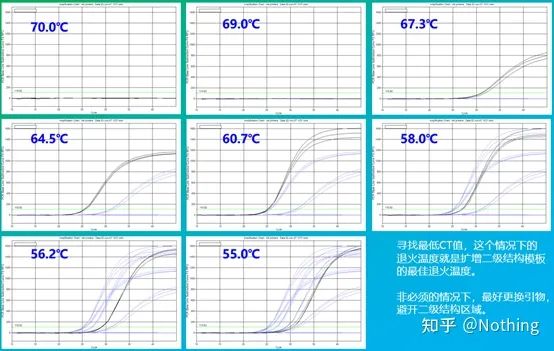
A estrutura secundária emerge
Pode-se observar que conforme o gradiente de temperatura diminui, os produtos começam a aparecer e o valor de Ct avança, atingindo o valor mínimo em 60,7°C, e então conforme o gradiente de temperatura diminui, o valor de Ct torna-se maior.Por outro lado, à medida que a temperatura aumenta, a estrutura secundária se abre e a eficiência de amplificação aumenta.Depois de atingir uma certa temperatura, aumentar a temperatura não pode melhorar a eficiência da amplificação.Porque os primers não podem ser combinados de forma estável neste momento.Portanto,procure a temperatura com o menor valor de Ct, que é a melhor temperatura para amplificar o gabarito da estrutura secundária!Claro, os espertinhos devem saber que se não for necessário, o melhor é trocar os primers e evitar a região da estrutura secundária.
5. Nível de aplicação
MIQE—Análise de Dados

A análise de dados é dada principalmente pelo instrumento PCR quantitativo fluorescente.No artigo anterior, muito trabalho de análise de dados foi feito, como o controle em branco, que foi explicado no projeto do experimento.Os genes de referência internos, números repetidos, etc. foram esclarecidos., aqui explicamos principalmente a aplicação do qPCR.
O qPCR é amplamente utilizado, e a verificação experimental e o diagnóstico de ácido nucleico são os cenários mais comumente usados.
quantificação absoluta
Log (concentração inicial) tem uma relação linear com o número de ciclos.Uma curva padrão pode ser desenhada a partir de um padrão com número de cópias inicial conhecido, ou seja, pode-se obter a relação linear da reação de amplificação.De acordo com o valor Ct da amostra, a concentração na amostra pode ser calculada.A quantidade de modelos a incluir.
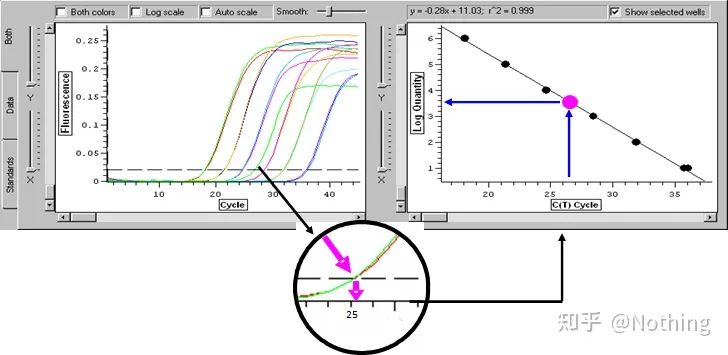
Método de Cálculo Quantitativo Absoluto
A quantificação absoluta deve ser baseada na curva padrão.Para fazer uma curva padrão, é necessário um padrão.Normalmente, o padrão é um plasmídeo obtido pela clonagem do gene alvo.Por que é um plasmídeo?Porque o DNA plasmidial circular é o mais estável.Dilua o produto padrão em 5 a 6 gradientes de acordo com a proporção de duplicação (diluição de 10 vezes) e preste atenção à uniformidade ao diluir.Deixe o valor Ct cair entre 15-30.
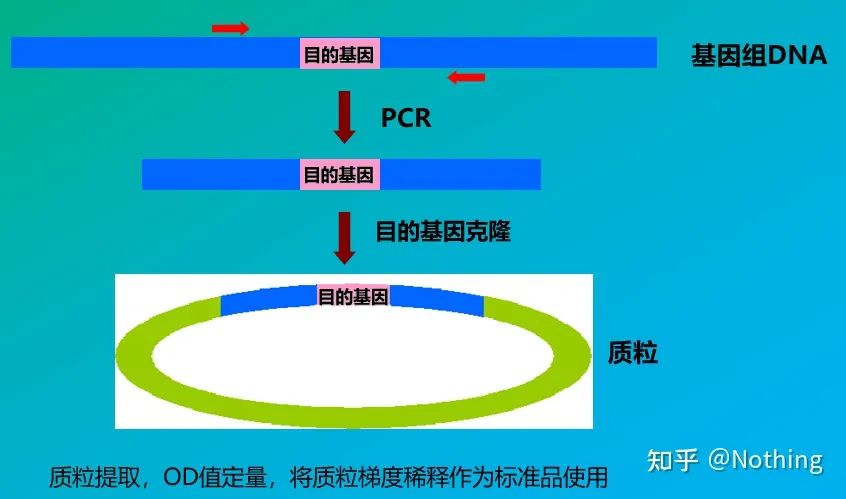
Preparação padrão
Ao mesmo tempo, a amostra a ser testada também deve ser diluída de acordo (lembre-se do fator de diluição) e o valor de Ct também deve estar entre 15-30.O produto padrão + a amostra a ser testada são colocados juntos na máquina.Após a corrida, uma curva padrão foi feita com a substância padrão e as amostras a serem testadas foram trazidas para a curva padrão para calcular a concentração.
A quantificação do VHB do vírus da hepatite B é uma quantificação absoluta típica, que pode calcular o número de cópias do vírus em 1ml de sangue.
Cálculo do número de cópias
Concentração da amostra a ser testada (ng/ul) = OD260 × 50ug/ml × fator de diluição
Peso molecular da amostra = número de bases × 324
O número de cópias da amostra a ser testada (cópias/ul) = a concentração da amostra a ser testada / o peso molecular da amostra × 6 × 1014

Método de cálculo do número de cópias
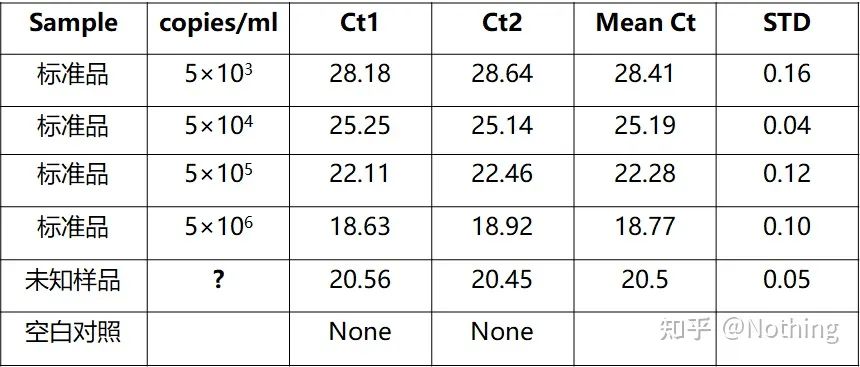

O acima é o método de cálculo para determinar a quantidade.Este é um problema matemático que pode ser resolvido depois de terminar o ensino médio, e os problemas matemáticos geralmente são resolvidos por computadores.Se você não entender, pode vir se comunicar.
quantificação relativa
A quantificação relativa é usada principalmente em pesquisas científicas.Quantos vírus existem em 1ml de sangue, e é um vírus de DNA, este é um evento relativamente determinístico: a quantidade de sangue pode ser determinada e o vírus de DNA é relativamente estável.No entanto, é difícil para nós comparar o número de cópias de transcrição de um determinado gene em uma folha, porque é difícil determinar o tamanho, peso e maciez da folha, a quantidade de RNA extraído é difícil de determinar e a eficiência da transcrição reversa também é difícil de determinar, ou seja, qualquer etapa pode fazer com que os dados experimentais tenham erros e não possam ser usados.
Portanto, a quantificação relativa deve introduzir um elemento:o gene de referência interna.
Em outras palavras, a quantificação relativa é, na verdade, uma comparação entre o gene alvo e o gene de referência interno.Comparado no mesmo tecido e na mesma célula, a influência do tamanho da amostra, quantidade de extração de RNA, eficiência de transcrição reversa e eficiência de PCR é relativamente pequena.Devido ao pequeno tamanho da amostra, tanto os genes de referência internos quanto os genes-alvo foram relativamente reduzidos.É por isso que enfatizamos a uniformidade e a estabilidade antes.
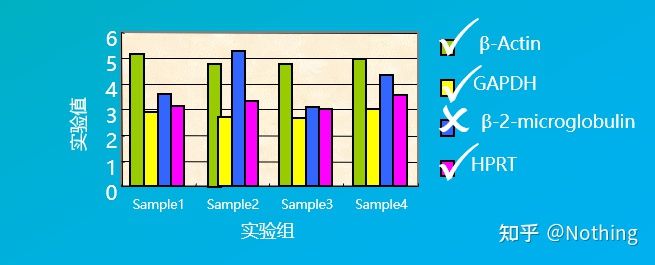
Genes de referência interna são geralmentegenes de limpeza(genes de manutenção), que se referem a uma classe de genes que são expressos de forma estável em todas as células, e seus produtos são necessários para manter as atividades vitais básicas das células.
Não confunda este conceito.Os genes de manutenção são termos de função biológica, enquanto os genes de referência interna são termos técnicos experimentais.Os genes de manutenção precisam passar pela validação antes de serem selecionados como genes de referência interna.
Por exemplo, selecionamos vários genes de manutenção na figura abaixo para testar seus níveis de expressão em diferentes células de tecidos e descobrimos que os níveis de expressão de β-2-microglobulina eram bastante diferentes dos outros três genes, portanto, não poderiam ser usados como genes de referência interna.
Depois de entender a função de correção do gene de referência interna, dois algoritmos são derivados devido à introdução do gene de referência interna.
·método de curva padrão duplo
·2 – Método △△Ct (método de comparação de valor CT)
Se você está interessado em estudar espécies e funções genéticas, por favor, desista da pesquisa sobre algoritmos e use fórmulas diretamente, ou use máquinas diretamente;se você é um cara hetero em matemática e engenharia, fique à vontade.
método da curva padrão duplo
Quantifique o gene alvo e o gene de manutenção da amostra de controle e da amostra a ser testada através da curva padrão e, em seguida, calcule o valor relativo de acordo com a fórmula de cálculo, que é o nível de expressão relativa.
Vantagens: análise simples, otimização experimental relativamente simples
Desvantagem: Para cada gene, cada rodada de experimentos deve fazer uma curva padrão
Aplicação: Um dos dois métodos quantitativos relativos mais comumente usados e reconhecidos no estudo da regulação da expressão gênica
A fórmula é a seguinte:

Os exemplos são os seguintes:
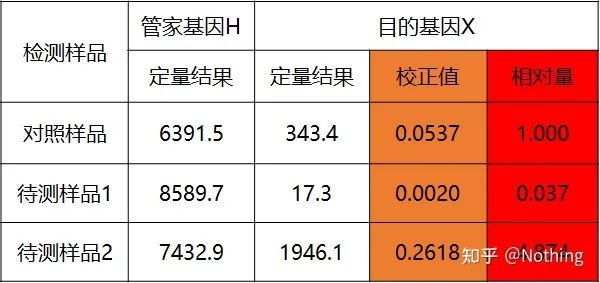
Calcule a quantidade relativa com base no resultado quantitativo
2 – Método △△Ct (método de comparação de valores CT)

Vantagens: Não há necessidade de fazer uma curva padrão
Desvantagens: Assume-se que a eficiência de amplificação está próxima de 100%;o desvio padrão é < 5%, e a curva padrão e a eficiência entre cada amplificação são consideradas consistentes;a otimização das condições experimentais é mais complicada.
Aplicação: Um dos dois métodos quantitativos relativos mais comumente usados e reconhecidos no estudo da regulação da expressão gênica
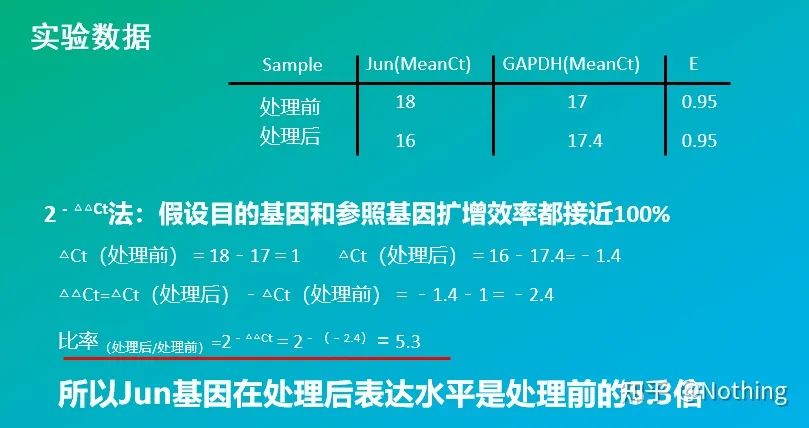
Claro, a eficiência de amplificação é geralmente impossível de ser perfeitamente 1. Método de correção: Se sabemos que o gene alvo e o gene de referência têm a mesma eficiência de amplificação, mas a eficiência de amplificação não é igual a 1, então 2-△△Ct pode ser corrigido como: (1+E )-△△Ct, por exemplo, se a eficiência de amplificação for 0,95, então a fórmula de cálculo pode ser corrigida para 1,95- △△Ct
Até agora, o conteúdo sobre PCR quantitativo fluorescente chegou ao fim.
Horário de postagem: 06 de abril de 2023



