



O RT-qPCR é desenvolvido a partir da tecnologia de PCR comum.Ele adiciona produtos químicos fluorescentes (corantes fluorescentes ou sondas fluorescentes) ao sistema de reação de PCR tradicional e detecta o processo de recozimento e extensão de PCR em tempo real de acordo com seus diferentes mecanismos luminescentes.As alterações do sinal fluorescente no meio são usadas para calcular a quantidade de alteração do produto em cada ciclo de PCR.Atualmente, os métodos mais comuns são o método do corante fluorescente e o método da sonda.
Método de corante fluorescente:
Alguns corantes fluorescentes, como SYBR Green Ⅰ, PicoGreen, BEBO, etc., não emitem luz por si mesmos, mas emitem fluorescência após a ligação ao sulco menor do dsDNA.Portanto, no início da reação de PCR, a máquina não consegue detectar o sinal fluorescente.Quando a reação prossegue para a extensão de recozimento (método de duas etapas) ou estágio de extensão (método de três etapas), as fitas duplas são abertas neste momento e a nova DNA polimerase.À medida que o número de ciclos de PCR aumenta, mais e mais corantes se combinam com o dsDNA, e o sinal fluorescente também é continuamente aprimorado.Tome SYBR Green Ⅰ como exemplo.
Método de sondagem:
A sonda Taqman é a sonda de hidrólise mais comumente usada.Há um grupo fluorescente na extremidade 5' da sonda, geralmente FAM.A própria sonda é uma sequência complementar ao gene alvo.Há um grupo de extinção fluorescente na extremidade 3' do fluoróforo.De acordo com o princípio da transferência de energia de ressonância de fluorescência (transferência de energia de ressonância de Förster, FRET), quando o grupo fluorescente repórter (molécula fluorescente doadora) e o grupo fluorescente de extinção (molécula fluorescente receptora) Quando o espectro de excitação se sobrepõe e a distância é muito próxima (7-10 nm), a excitação da molécula doadora pode induzir a fluorescência da molécula receptora, enquanto a autofluorescência é enfraquecida.Portanto, no início da reação de PCR, quando a sonda estiver livre e intacta no sistema, o grupo fluorescente repórter não emitirá fluorescência.Ao recozimento, o primer e a sonda se ligam ao molde.Durante o estágio de extensão, a polimerase sintetiza continuamente novas cadeias.A DNA polimerase tem atividade exonuclease 5'-3'.Ao atingir a sonda, a DNA polimerase irá hidrolisar a sonda do molde, separar o grupo fluorescente repórter do grupo fluorescente supressor e liberar o sinal fluorescente.Como existe uma relação de um para um entre a sonda e o modelo, o método da sonda é superior ao método do corante em termos de precisão e sensibilidade do teste.
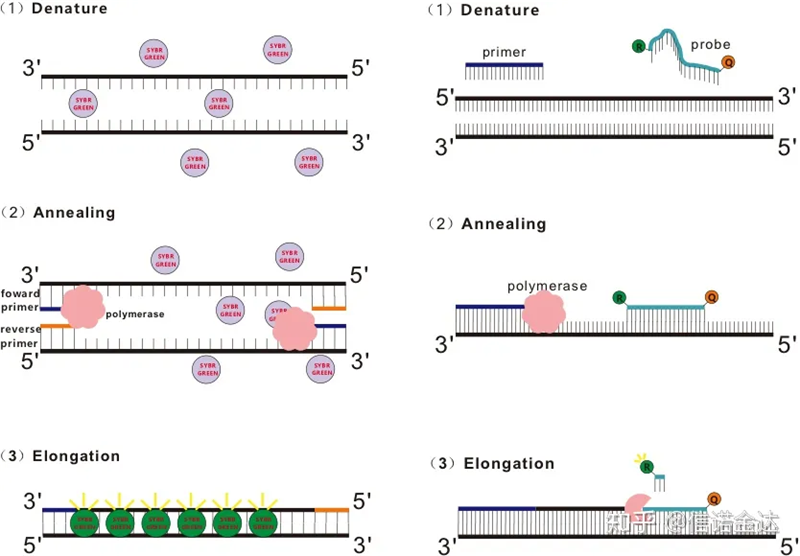
Fig. 1 Princípio de qRT-PCR
Projeto de primer
Princípios:
Os primers devem ser desenhados na região conservada da série de ácidos nucléicos e ter especificidade.
É melhor usar a sequência de cDNA, e a sequência de mRNA também é aceitável.Caso contrário, descubra o design da região cds da sequência de DNA.
O comprimento do produto quantitativo fluorescente é 80-150bp, o mais longo é 300bp, o comprimento do primer é geralmente entre 17-25 bases, e a diferença entre os primers upstream e downstream não deve ser muito grande.
O conteúdo G+C está entre 40% e 60%, e 45-55% é o melhor.
O valor TM está entre 58-62 graus.
Tente evitar primer dímeros e autodímeros, (não aparecem mais de 4 pares de bases complementares consecutivas) estrutura em grampo, se inevitável, faça ΔG<4.5kJ/mol* Se você não puder garantir que o gDNA foi removido durante a transcrição reversa Limpo, é melhor projetar os primers do íntron
específica A homologia da sequência amplificada heterogeneamente é preferencialmente inferior a 70% ou tem homologia de 8 bases complementares.
Base de dados:
Pesquisa CottonFGD por palavras-chave
Projeto primário:
Projeto de primer IDT-qPCR
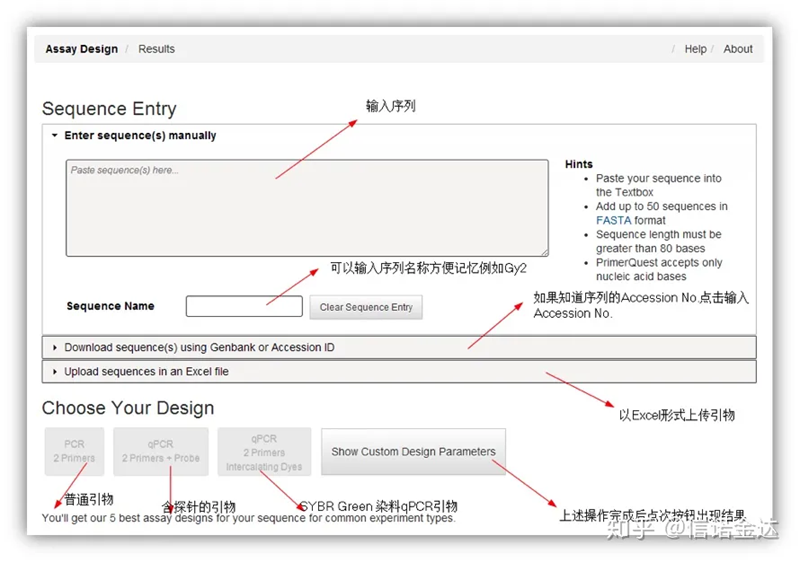
Página da ferramenta de design de primer on-line Fig2 IDT
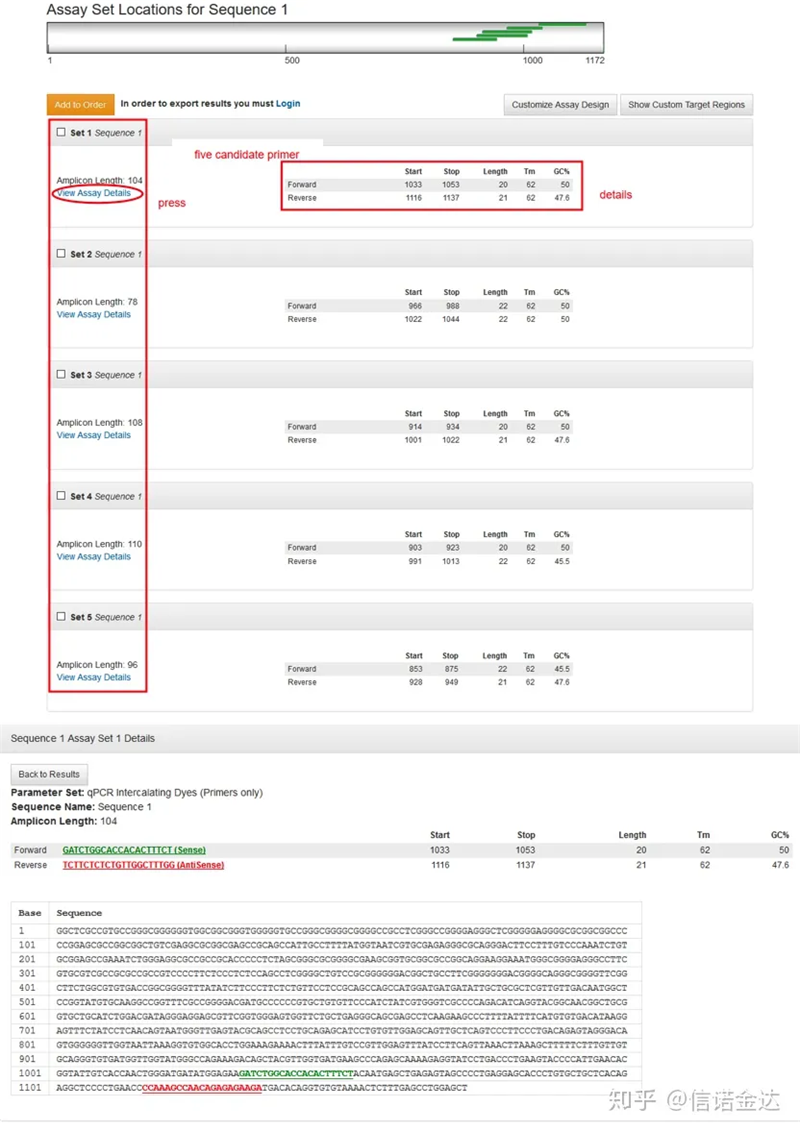
Fig3 exibição da página de resultados
Projeto de primers lncRNA:
lncRNA:as mesmas etapas do mRNA.
miRNA:O princípio do método stem-loop: como todos os miRNAs são sequências curtas de cerca de 23 nt, a detecção direta por PCR não pode ser realizada, portanto, a ferramenta de sequência stem-loop é usada.A sequência de haste-alça é um DNA de fita simples de cerca de 50 nt, que pode formar uma estrutura em grampo por si só.3 'A extremidade pode ser projetada como uma sequência complementar ao fragmento parcial de miRNA, então o miRNA alvo pode ser conectado à sequência haste-loop durante a transcrição reversa, e o comprimento total pode atingir 70bp, o que está alinhado com o comprimento do produto amplificado determinado por qPCR.Projeto de iniciador de miRNA de cauda.
Detecção específica de amplificação:
Banco de dados de detonação online: detonação CottonFGD por similaridade de sequência
Explosão local: Consulte o uso do Blast+ para fazer explosão local, linux e macos podem estabelecer diretamente um banco de dados local, o sistema win10 também pode ser feito após a instalação do ubuntu bash.Criar banco de dados de explosão local e explosão local;abra o ubuntu bash no win10.
Aviso: O algodão de terras altas e o algodão de ilhas marinhas são culturas tetraploides, portanto, o resultado da explosão geralmente será de dois ou mais fósforos.No passado, o uso de NAU cds como banco de dados para realizar o blast provavelmente encontraria dois genes homólogos com apenas algumas diferenças de SNP.Normalmente, os dois genes homólogos não podem ser separados por design de primer, então eles são tratados como iguais.Se houver um indel óbvio, o primer geralmente é projetado no indel, mas isso pode levar à estrutura secundária do primer. A energia livre torna-se mais alta, levando a uma diminuição na eficiência da amplificação, mas isso é inevitável.
Detecção da estrutura secundária do primer:
Passos:abrir oligo 7 → inserir a sequência do modelo → fechar a subjanela → salvar → localizar o primer no modelo, pressionar ctrl+D para definir o comprimento do primer → analisar várias estruturas secundárias, como corpo de autodimerização, heterodímero, hairpin, incompatibilidade, etc. As duas últimas imagens na Figura 4 são os resultados do teste dos primers.O resultado do primer frontal é bom, não há estrutura óbvia de dímero e grampo de cabelo, nenhuma base complementar contínua e o valor absoluto da energia livre é inferior a 4,5, enquanto o primer traseiro mostra contínuo As 6 bases são complementares e a energia livre é 8,8;além disso, um dímero mais sério aparece na extremidade 3' e um dímero de 4 bases consecutivas aparece.Embora a energia livre não seja alta, o dímero 3' Chl pode afetar seriamente a especificidade e a eficiência da amplificação.Além disso, é necessário verificar grampos de cabelo, heterodímeros e incompatibilidades.
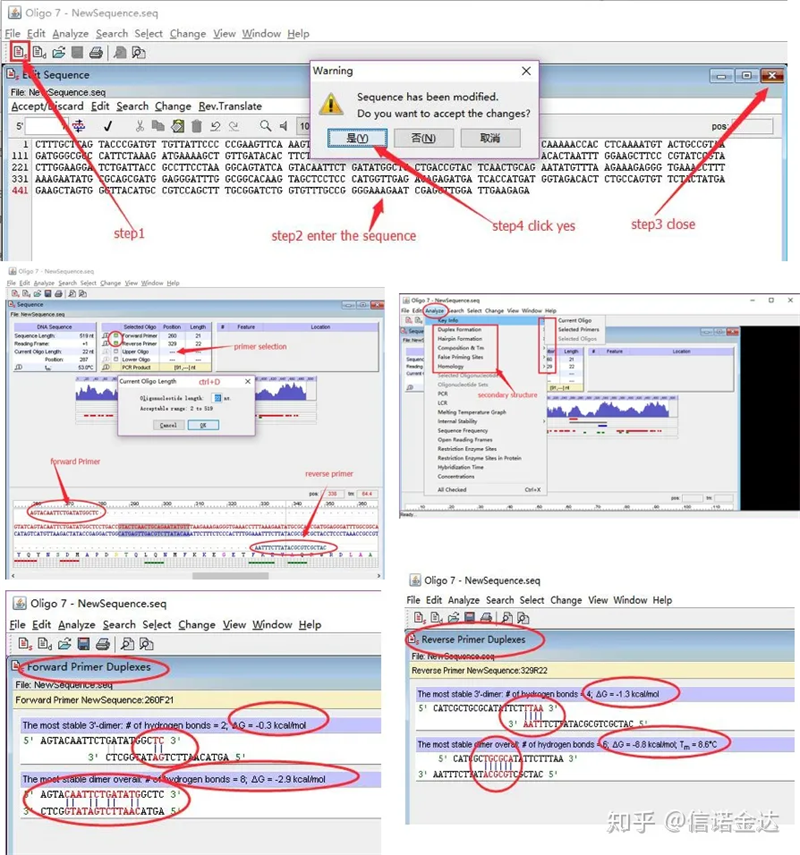
Resultados da detecção de oligo7 da Fig3
Detecção de eficiência de amplificação:
A eficiência de amplificação da reação de PCR afeta seriamente os resultados de PCR.Também em qRT-PCR, a eficiência de amplificação é particularmente importante para os resultados quantitativos.Remova outras substâncias, máquinas e protocolos no tampão de reação.A qualidade dos primers também tem grande influência na eficiência de amplificação do qRT-PCR.Para garantir a precisão dos resultados, tanto a quantificação de fluorescência relativa quanto a quantificação de fluorescência absoluta precisam detectar a eficiência de amplificação dos primers.Reconhece-se que a eficiência efetiva da amplificação qRT-PCR está entre 85% e 115%.Existem dois métodos:
1. Método da curva padrão:
a.Misturar cDNA
b.diluição gradiente
c.qPCR
d.Equação de regressão linear para calcular a eficiência de amplificação
2. LinRegPCR
LinRegPCR é um programa para a análise de dados RT-PCR em tempo real, também chamados de dados quantitativos de PCR (qPCR) baseados em SYBR Green ou química similar.O programa usa dados corrigidos sem linha de base, executa uma correção de linha de base em cada amostra separadamente, determina uma janela de linearidade e então usa análise de regressão linear para ajustar uma linha reta através do conjunto de dados de PCR.A partir da inclinação desta linha é calculada a eficiência de PCR de cada amostra individual.A eficiência média de PCR por amplicon e o valor Ct por amostra são usados para calcular uma concentração inicial por amostra, expressa em unidades de fluorescência arbitrárias.A entrada e saída de dados são feitas através de uma planilha Excel.Apenas amostra
mistura é necessária, sem gradiente
passos são necessários:(Tome o Bole CFX96 como exemplo, não exatamente Máquina com ABI claro)
experimentar:é um experimento qPCR padrão.
saída de dados qPCR:LinRegPCR pode reconhecer duas formas de arquivos de saída: RDML ou quantificação Resultado da amplificação.Na verdade, é o valor de detecção em tempo real do número do ciclo e do sinal de fluorescência pela máquina, e a amplificação é obtida analisando o valor da mudança de fluorescência da eficiência do segmento linear.
Seleção de dados: em teoria, o valor RDML deve ser utilizável.Estima-se que o problema do meu computador seja que o software não reconhece RDML, portanto, tenho o valor de saída do Excel como os dados originais.Recomenda-se realizar uma triagem aproximada dos dados primeiro, como falha na adição de amostras, etc. Os pontos podem ser excluídos nos dados de saída (claro, você não pode excluí-los, o LinRegPCR irá ignorar esses pontos no estágio posterior)
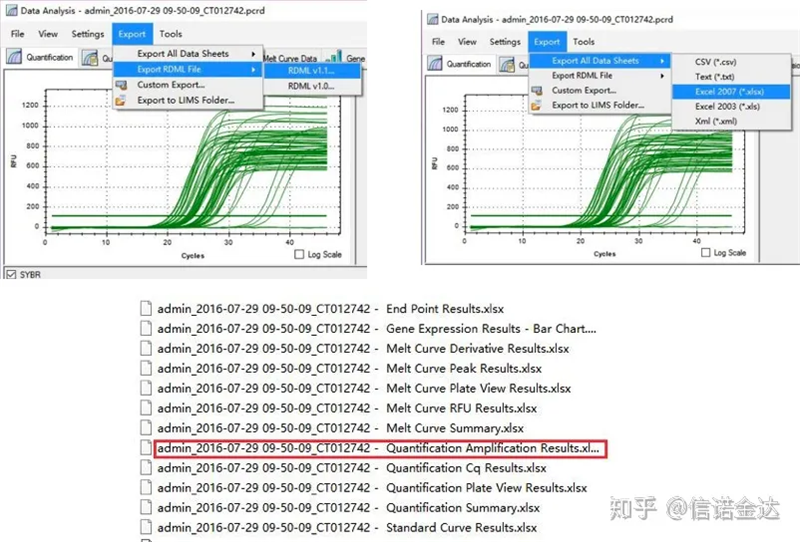
Fig5 exportação de dados qPCR
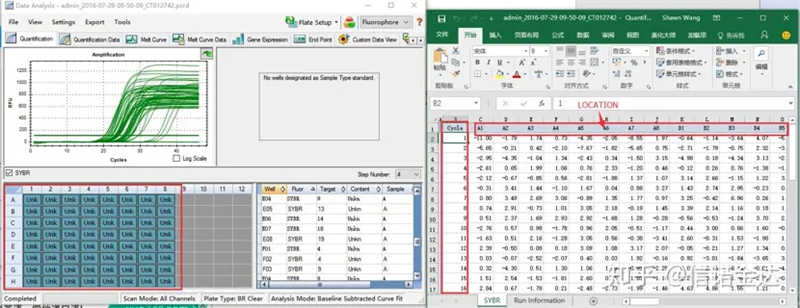
Fig6 seleção de amostras candidatas
Entrada de dados:Abra os resultados da amplificação de qualificação.xls, → abra LinRegPCR → arquivo → leia do Excel → selecione os parâmetros conforme mostrado na Figura 7 → OK → clique em determinar linhas de base
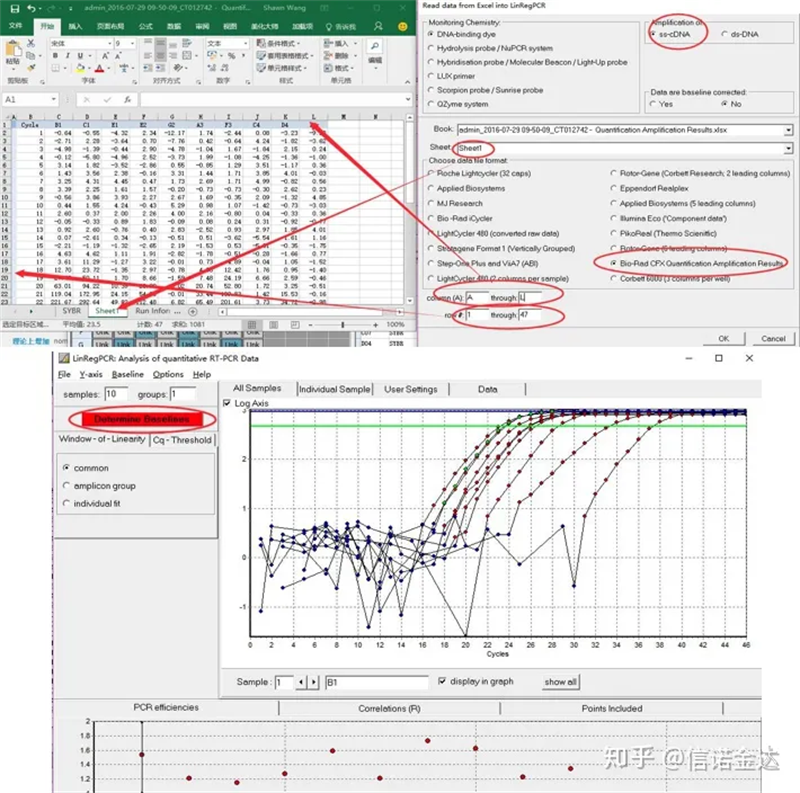
Fig7 etapas da entrada de dados linRegPCR
Resultado:Se não houver repetição, nenhum agrupamento é necessário.Caso haja repetição, o agrupamento pode ser editado no agrupamento amostral, e o nome do gene é inserido no identificador, e então o mesmo gene será automaticamente agrupado.Por fim, clique no arquivo, exporte o Excel e visualize os resultados.A eficiência de amplificação e os resultados R2 de cada poço serão exibidos.Em segundo lugar, se você dividir em grupos, a eficiência de amplificação média corrigida será exibida.Certifique-se de que a eficiência de amplificação de cada primer esteja entre 85% e 115%.Se for muito grande ou muito pequeno, significa que a eficiência de amplificação do primer é ruim.
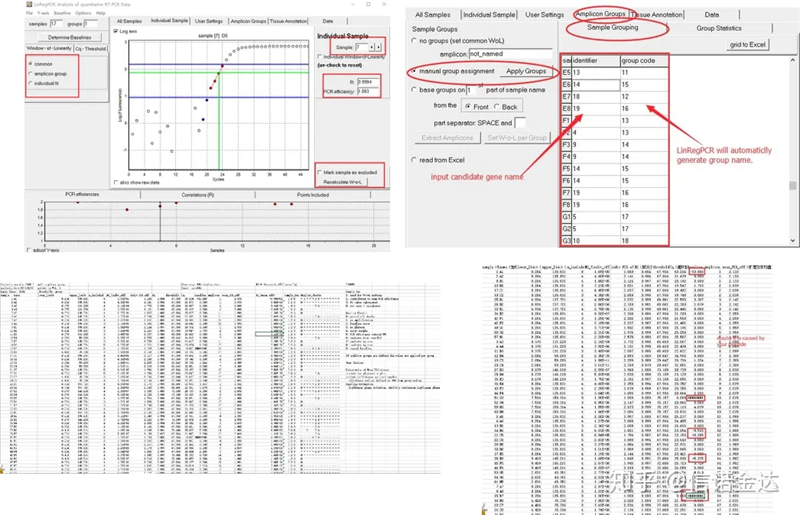
Fig 8 Resultado e saída de dados
Processo experimental:
Requisitos de qualidade de RNA:
Pureza:1.72.0 indica que pode haver isotiocianato residual.O ácido nucleico limpo A260/A230 deve estar em torno de 2 . Se houver uma forte absorção em 230 nm, isso indica que existem compostos orgânicos, como íons fenato.Além disso, pode ser detectado por eletroforese em gel de agarose a 1,5%.Aponte o marcador, porque o ssRNA não tem desnaturação e o logaritmo do peso molecular não tem uma relação linear, e o peso molecular não pode ser expresso corretamente.Concentração: Teoricamentenãoinferior a 100 ng/ul, se a concentração for muito baixa, a pureza geralmente é baixa e não alta
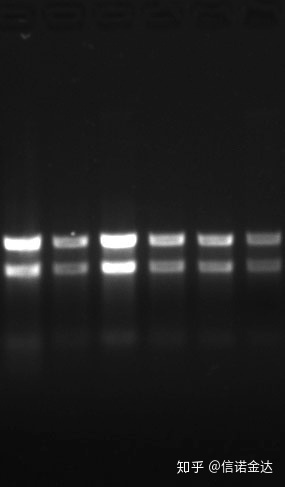
Gel de RNA Fig9
Além disso, se a amostra for preciosa e a concentração de RNA for alta, recomenda-se aliquotá-la após a extração e diluir o RNA até uma concentração final de 100-300 ng/ul para transcrição reversa.Emo processo de transcrição reversa, quando o mRNA é transcrito, primers oligo (dt) que podem se ligar especificamente a caudas poliA são usados para transcrição reversa, enquanto lncRNA e circRNA usam primers hexâmero aleatório (Random 6 mer) para transcrição reversa do RNA total Para miRNA, primers neck-loop específicos de miRNA são usados para transcrição reversa.Muitas empresas já lançaram kits especiais de rejeitos.Para o método de haste-laço, o método de cauda é mais conveniente, de alto rendimento e economia de reagente, mas o efeito de distinguir miRNAs da mesma família não deve ser tão bom quanto o método de haste-laço.Cada kit de transcrição reversa tem requisitos para a concentração de primers específicos do gene (stem-loops).A referência interna usada para miRNA é U6.No processo de inversão haste-loop, um tubo de U6 deve ser invertido separadamente, e os primers dianteiro e traseiro de U6 devem ser adicionados diretamente.Tanto o circRNA quanto o lncRNA podem usar HKGs como referência interna.Emdetecção de cDNA,
se não houver problema com o RNA, o cDNA também deve funcionar.No entanto, se a perfeição do experimento for perseguida, é melhor usar um gene de referência interno (gene de referência, RG) que possa distinguir gDNA de cds.Geralmente, RG é um gene de limpeza., HKG) conforme mostrado na Figura 10;Naquela época, eu estava fazendo proteína de armazenamento de soja e usava íntrons contendo actina 7 como referência interna.O tamanho do fragmento amplificado deste primer em gDNA era de 452 pb, e se o cDNA fosse usado como molde, era de 142 pb.Em seguida, os resultados do teste descobriram que parte do cDNA estava realmente contaminado pelo gDNA e também provou que não havia problema com o resultado da transcrição reversa e que poderia ser usado como modelo para PCR.É inútil executar eletroforese em gel de agarose diretamente com cDNA, e é uma banda difusa, que não é convincente.
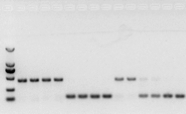
Fig. 10 Detecção de cDNA
A determinação das condições de qPCRgeralmente não há problema de acordo com o protocolo do kit, principalmente na etapa do valor tm.Se alguns primers não forem bem projetados durante o design do primer, resultando em uma grande diferença entre o valor tm e os 60°C teóricos, é recomendável que o cDNA Após as amostras serem misturadas, execute um PCR gradiente com primers e tente evitar definir a temperatura sem bandas como o valor TM.
Análise de dados
O método convencional de processamento de PCR quantitativo de fluorescência relativa é basicamente de acordo com 2-ΔΔCT.Modelo de processamento de dados.
Produtos relacionados:
PCR em tempo real fácilTM –Taqman
PCR em tempo real fácilTM –SYBR VERDE I
RT Easy I (Master Premix para a síntese da primeira fita de cDNA)
RT Easy II (Master Premix para síntese de primeira fita de cDNA para qPCR)
Horário da postagem: 14 de março de 2023



