



A PCR (reação em cadeia da polimerase) é uma das tecnologias de amplificação de DNA in vitro, com uma história de mais de 30 anos.
A tecnologia de PCR foi pioneira por Kary Mullis de Cetus, EUA, em 1983. Mullis solicitou uma patente de PCR em 1985 e publicou o primeiro artigo acadêmico sobre PCR no Science no mesmo ano.Mullis recebeu o Prêmio Nobel de Química em 1993 por seu trabalho.
Princípios Básicos da PCR
A PCR pode amplificar fragmentos de DNA alvo em mais de um milhão de vezes.O princípio está sob a catálise da DNA polimerase, usando o DNA da fita parental como molde e primer específico como ponto de partida para a extensão.É replicado in vitro por meio de etapas como desnaturação, recozimento e extensão.O processo de DNA de fita filha complementar ao DNA modelo de fita pai.

O processo de PCR padrão é dividido em três etapas:
1. Desnaturação: Use alta temperatura para separar as fitas duplas de DNA.A ponte de hidrogênio entre as fitas duplas do DNA é quebrada em alta temperatura (93-98 ℃).
2.Recozimento: Após a separação do DNA de fita dupla, abaixe a temperatura para que o primer possa se ligar ao DNA de fita simples.
3. Extensão: A DNA polimerase começa a sintetizar filamentos complementares ao longo dos filamentos de DNA a partir dos primers ligados quando a temperatura é reduzida.Quando a extensão é concluída, um ciclo é concluído e o número de fragmentos de DNA dobra.
Alternando essas três etapas 25 a 35 vezes, o número de fragmentos de DNA aumentará exponencialmente.
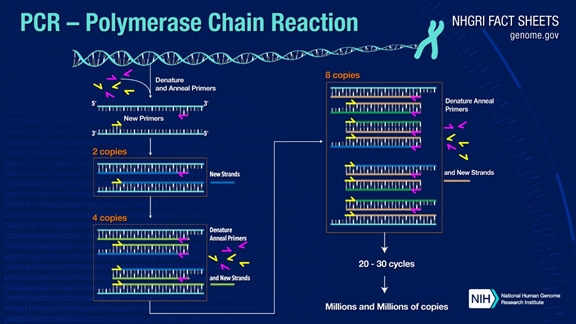
A engenhosidade da PCR é que diferentes iniciadores podem ser projetados para diferentes genes-alvo, de modo que os fragmentos do gene-alvo possam ser amplificados em um curto período de tempo.
Até agora, a PCR pode ser dividida em três categorias: PCR comum, PCR quantitativo fluorescente e PCR digital.
A primeira geração de PCR comum
Use um instrumento de amplificação de PCR comum para amplificar o gene alvo e, em seguida, use a eletroforese em gel de agarose para detectar o produto, apenas a análise qualitativa pode ser feita.
As principais desvantagens do PCR de primeira geração:
1.Propenso a amplificação não específica e resultados falsos positivos.
2.A detecção leva muito tempo e a operação é complicada.
3. Somente teste qualitativo pode ser feito
PCR em tempo real de segunda geração
A PCR em tempo real, também conhecida como qPCR, utiliza sondas fluorescentes que podem indicar o andamento do sistema de reação e monitora o acúmulo de produtos amplificados por meio do acúmulo de sinais fluorescentes e julga os resultados pela curva de fluorescência.Pode ser quantificado com a ajuda do valor Cq e da curva padrão.
Como a tecnologia qPCR é realizada em sistema fechado, a probabilidade de contaminação é reduzida e o sinal de fluorescência pode ser monitorado para detecção quantitativa, por isso é a mais utilizada na prática clínica e tornou-se a tecnologia dominante em PCR.
As substâncias fluorescentes usadas na PCR quantitativa fluorescente em tempo real podem ser divididas em: sonda fluorescente TaqMan, faróis moleculares e corante fluorescente.
1) Sonda fluorescente TaqMan:
Durante a amplificação por PCR, uma sonda fluorescente específica é adicionada ao adicionar um par de primers.A sonda é um oligonucleotídeo e ambas as extremidades são marcadas com um grupo fluorescente repórter e um grupo fluorescente extintor.
Quando a sonda está intacta, o sinal fluorescente emitido pelo grupo repórter é absorvido pelo grupo quenching;durante a amplificação por PCR, a atividade da exonuclease 5'-3' da enzima Taq cliva e degrada a sonda, tornando o grupo fluorescente repórter e extintor O grupo fluorescente é separado, para que o sistema de monitoramento de fluorescência possa receber o sinal de fluorescência, ou seja, toda vez que uma fita de DNA é amplificada, uma molécula fluorescente é formada e o acúmulo do sinal de fluorescência é completamente sincronizado com a formação do produto de PCR.
2) Corante fluorescente SYBR:
No sistema de reação de PCR, um excesso de corante fluorescente SYBR é adicionado.Depois que o corante fluorescente SYBR é incorporado não especificamente na fita dupla do DNA, ele emite um sinal fluorescente.A molécula de corante SYBR que não for incorporada à cadeia não emitirá nenhum sinal fluorescente, garantindo assim o sinal fluorescente. O aumento dos produtos de PCR é completamente sincronizado com o aumento dos produtos de PCR.O SYBR se liga apenas ao DNA de fita dupla, portanto, a curva de fusão pode ser usada para determinar se a reação de PCR é específica.

3) Farol molecular:
É uma sonda oligonucleotídica duplamente marcada com haste-alça que forma uma estrutura em grampo de cerca de 8 bases nas extremidades 5 e 3.As sequências de ácidos nucléicos em ambas as extremidades são emparelhadas complementarmente, fazendo com que o grupo fluorescente e o grupo de extinção sejam rígidos.Perto, nenhuma fluorescência será produzida.

Depois que o produto de PCR é gerado, durante o processo de recozimento, a parte do meio do farol molecular é emparelhada com uma sequência de DNA específica e o gene fluorescente é separado do gene supressor para produzir fluorescência.

As principais desvantagens do PCR de segunda geração:
Ainda falta sensibilidade e a detecção de espécimes de baixa cópia é imprecisa.
Existe a influência do valor de fundo e o resultado é suscetível a interferências.
Quando há inibidores de PCR no sistema de reação, os resultados da detecção são suscetíveis a interferências.
PCR digital de terceira geração
A PCR digital (DigitalPCR, dPCR, Dig-PCR) calcula o número de cópias da sequência alvo por meio da detecção de ponto final e pode realizar detecção quantitativa absoluta precisa sem usar controles internos e curvas padrão.
A PCR digital usa detecção de ponto final e não depende do valor Ct (limiar do ciclo), portanto a reação da PCR digital é menos afetada pela eficiência da amplificação e a tolerância aos inibidores da reação da PCR é aprimorada, com alta precisão e reprodutibilidade.
Devido às características de alta sensibilidade e alta precisão, não é facilmente interferido por inibidores de reação de PCR e pode alcançar a verdadeira quantificação absoluta sem produtos padrão, que se tornou um hotspot de pesquisa e aplicação.
De acordo com as diferentes formas da unidade de reação, ela pode ser dividida em três tipos principais: sistemas microfluídicos, chip e gotículas.
1) PCR digital microfluídica, mdPCR:
Com base na tecnologia microfluídica, o modelo de DNA é separado.A tecnologia microfluídica pode realizar a nano-atualização da amostra ou a geração de gotículas menores, mas as gotículas precisam de um método de adsorção especial e, em seguida, combinadas com o sistema de reação de PCR.O mdPCR foi gradualmente adotado por outros métodos de substituição.
2) PCR digital baseado em gotículas, ddPCR:
Use a tecnologia de geração de gotículas de água em óleo para processar a amostra em gotículas e divida o sistema de reação contendo moléculas de ácido nucleico em milhares de gotículas em nanoescala, cada uma das quais não contém a molécula alvo de ácido nucleico a ser detectada ou contém uma a várias moléculas alvo de ácido nucleico a serem testadas.
3) PCR digital baseado em chip, cdPCR:
Use a tecnologia de via de fluido integrada para gravar muitos microtubos e microcavidades em pastilhas de silício ou vidro de quartzo e controlar o fluxo da solução através de diferentes válvulas de controle e dividir o líquido da amostra em nanômetros do mesmo tamanho nos poços de reação para reação de PCR digital para obter quantificação absoluta.
As principais desvantagens do PCR de terceira geração:
Os equipamentos e reagentes são caros.
Os requisitos de qualidade do modelo são altos.Se a quantidade do modelo exceder a quantidade do microssistema, será impossível quantificar e, se for muito pequena, a precisão da quantificação será reduzida.
Falsos positivos também podem ser gerados quando há amplificação inespecífica.
Horário da postagem: 30 de julho de 2021



