



- PCR é um método usado para amplificar DNA a partir de uma pequena quantidade de modelo de DNA.A RT-PCR utiliza transcrição reversa para produzir um modelo de DNA a partir de uma fonte de RNA que pode então ser amplificado.
- PCR e RT-PCR são normalmente reações de ponto final, enquanto qPCR e RT-qPCR usam a cinética da taxa de síntese do produto durante a reação de PCR para quantificar a quantidade de modelo presente.
- Métodos mais recentes, como a PCR digital, fornecem quantificação absoluta do modelo inicial de DNA, enquanto métodos como a PCR isotérmica reduzem a necessidade de equipamentos caros para fornecer resultados confiáveis.
A reação em cadeia da polimerase (PCR) é uma técnica de biologia molecular relativamente simples e amplamente utilizada para amplificar e detectar sequências de DNA e RNA.Em comparação com os métodos tradicionais de clonagem e amplificação de DNA, que muitas vezes podem levar dias, a PCR requer apenas algumas horas.A PCR é altamente sensível e requer um modelo mínimo para detecção e amplificação de sequências específicas.Os métodos básicos de PCR avançaram ainda mais a partir da simples detecção de DNA e RNA.Abaixo, fornecemos uma visão geral dos diferentes métodos de PCR e dos reagentes que fornecemos na Enzo Life Sciences para suas necessidades de pesquisa.Nosso objetivo é ajudar os cientistas a acessar rapidamente os reagentes de PCR para usar em seu próximo projeto de pesquisa!
PCR
Para PCR padrão, tudo que você precisa é de uma DNA polimerase, magnésio, nucleotídeos, primers, o modelo de DNA a ser amplificado e um termociclador.O mecanismo de PCR é tão simples quanto seu propósito: 1) o DNA de fita dupla (dsDNA) é desnaturado pelo calor, 2) os primers se alinham às fitas simples de DNA e 3) os primers são estendidos pela DNA polimerase, resultando em duas cópias do fita original de DNA.O processo de desnaturação, recozimento e alongamento ao longo de uma série de temperaturas e tempos é conhecido como um ciclo de amplificação (Fig. 1).
|
|
| Figura 1.Representação esquemática de um ciclo de amplificação por PCR. |
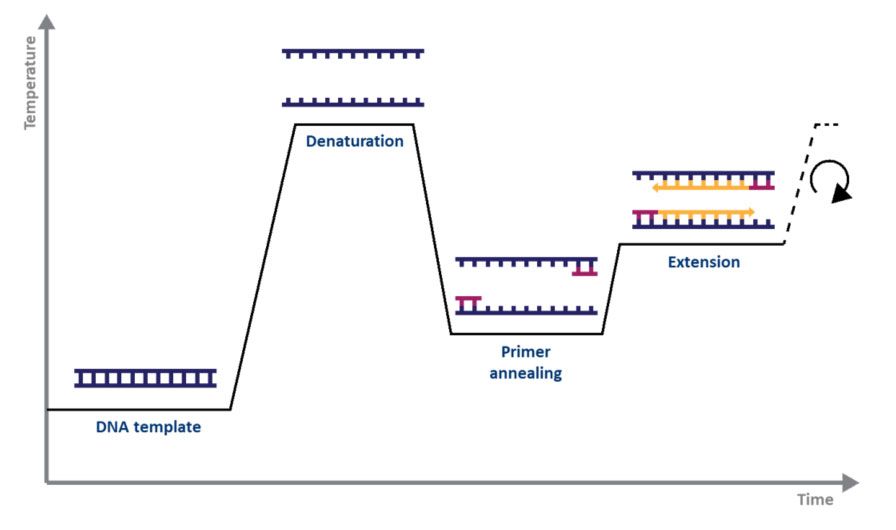
Cada etapa do ciclo deve ser otimizada para o modelo e conjunto de primers utilizados.Este ciclo é repetido aproximadamente 20-40 vezes, e o produto amplificado pode então ser analisado, normalmente por gel de agarose (Fig. 2).
| |
| Figura 2.Amplificação de um molde de DNA por PCR e análise por eletroforese em gel de agarose. |
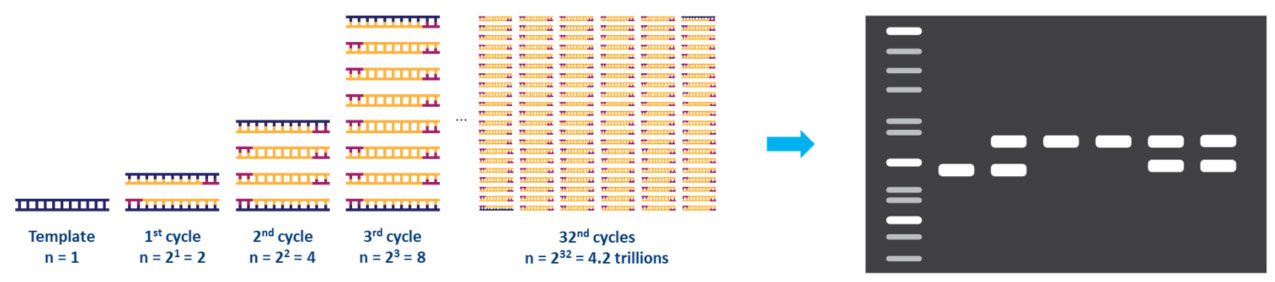
Como a PCR é um método altamente sensível e são necessários volumes muito pequenos para reações únicas, recomenda-se a preparação de uma mistura mestre para diversas reações.A mistura principal deve ser bem misturada e depois dividida pelo número de reações, garantindo que cada reação conterá a mesma quantidade de enzima, dNTPs e primers.Muitos fornecedores, como a Enzo Life Sciences, também oferecem misturas de PCR que já contêm tudo, exceto os primers e o modelo de DNA.
Regiões ricas em guanina/citosina (ricas em GC) representam um desafio nas técnicas padrão de PCR.As sequências ricas em GC são mais estáveis que as sequências com menor conteúdo de GC.Além disso, sequências ricas em GC tendem a formar estruturas secundárias, como alças em gancho.Como resultado, as cadeias duplas ricas em GC são difíceis de separar completamente durante a fase de desnaturação.Conseqüentemente, a DNA polimerase não consegue sintetizar a nova fita sem obstáculos.Uma temperatura de desnaturação mais alta pode melhorar isso, e ajustes para uma temperatura de recozimento mais alta e um tempo de recozimento mais curto podem impedir a ligação inespecífica de primers ricos em GC.Reagentes adicionais podem aumentar a amplificação de sequências ricas em GC.DMSO, glicerol e betaína ajudam a romper as estruturas secundárias causadas pelas interações de GC e, assim, facilitam a separação das fitas duplas.
PCR de início quente
A amplificação inespecífica é um problema que pode ocorrer durante a PCR.A maioria das DNA polimerases usadas para PCR funcionam melhor em temperaturas em torno de 68°C a 72°C.A enzima pode, no entanto, também ser activa a temperaturas mais baixas, embora num grau inferior.A temperaturas muito abaixo da temperatura de recozimento, os iniciadores podem ligar-se de forma não específica e conduzir a uma amplificação não específica, mesmo que a reação seja realizada em gelo.Isto pode ser evitado usando inibidores da polimerase que se dissociam da DNA polimerase apenas quando uma determinada temperatura é atingida, daí o termo PCR de início a quente.O inibidor pode ser um anticorpo que se liga à polimerase e desnatura à temperatura inicial de desnaturação (normalmente 95°C).
Polimerase de alta fidelidade
Embora as DNA polimerases amplifiquem com bastante precisão a sequência modelo original, podem ocorrer erros na correspondência de nucleotídeos.Incompatibilidades em aplicações como a clonagem podem resultar em transcrições truncadas e proteínas mal traduzidas ou inativas a jusante.Para evitar essas incompatibilidades, polimerases com atividade de “revisão” foram identificadas e incorporadas ao fluxo de trabalho.A primeira polimerase de revisão, Pfu, foi identificada em 1991 em Pyrococcus furiosus.Esta enzima Pfu tem atividade exonuclease de 3' a 5'.À medida que o DNA é amplificado, a exonuclease remove nucleotídeos incompatíveis na extremidade 3' da fita.O nucleotídeo correto é então substituído e a síntese de DNA continua.A identificação de sequências de nucleotídeos incorretas baseia-se na afinidade de ligação do nucleosídeo trifosfato correto com a enzima, onde a ligação ineficiente retarda a síntese e permite a substituição correta.A atividade de revisão da polimerase Pfu resulta em menos erros na sequência final em comparação com a DNA polimerase Taq.Nos últimos anos, outras enzimas de revisão foram identificadas e foram feitas modificações na enzima Pfu original para reduzir ainda mais a taxa de erro durante a amplificação do DNA.
RT-PCR
PCR de transcrição reversa, ou RT-PCR, permite o uso de RNA como modelo.Uma etapa adicional permite a detecção e amplificação de RNA.O RNA é transcrito reversamente em DNA complementar (cDNA), usando transcriptase reversa.A qualidade e pureza do modelo de RNA são essenciais para o sucesso do RT-PCR.A primeira etapa do RT-PCR é a síntese de um híbrido DNA/RNA.A transcriptase reversa também possui uma função RNase H, que degrada a porção de RNA do híbrido.A molécula de DNA de fita simples é então completada pela atividade da DNA polimerase dependente de DNA da transcriptase reversa em cDNA.A eficiência da reação da primeira fita pode afetar o processo de amplificação.A partir daqui, o procedimento padrão de PCR é utilizado para amplificar o cDNA.A possibilidade de reverter o RNA em cDNA por RT-PCR tem muitas vantagens e é usada principalmente para análise de expressão gênica.O RNA é de fita simples e muito instável, o que torna difícil trabalhar com ele.Geralmente serve como uma primeira etapa no qPCR, que quantifica transcritos de RNA em uma amostra biológica.
qPCR e RT-qPCR
A PCR quantitativa (qPCR) é usada para detectar, caracterizar e quantificar ácidos nucleicos para inúmeras aplicações.Na RT-qPCR, os transcritos de RNA são frequentemente quantificados pela transcrição reversa primeiro em cDNA, conforme descrito acima, e então o qPCR é posteriormente realizado.Tal como na PCR padrão, o DNA é amplificado por três etapas repetidas: desnaturação, recozimento e alongamento.No entanto, no qPCR, a marcação fluorescente permite a coleta de dados à medida que a PCR avança.Esta técnica tem muitos benefícios devido à variedade de métodos e produtos químicos disponíveis.
No qPCR baseado em corante (tipicamente verde), a marcação fluorescente permite a quantificação das moléculas de DNA amplificadas, empregando o uso de um corante de ligação ao dsDNA.Durante cada ciclo, a fluorescência é medida.O sinal de fluorescência aumenta proporcionalmente à quantidade de DNA replicado.Assim, o DNA é quantificado em “tempo real” (Fig. 3).As desvantagens do qPCR baseado em corante são que apenas um alvo pode ser examinado por vez e que o corante se ligará a qualquer ds-DNA presente na amostra.
|
|
| Figura 3.Amplificando um modelo de DNA por qPCR e medindo o sinal de fluorescência em tempo real. |
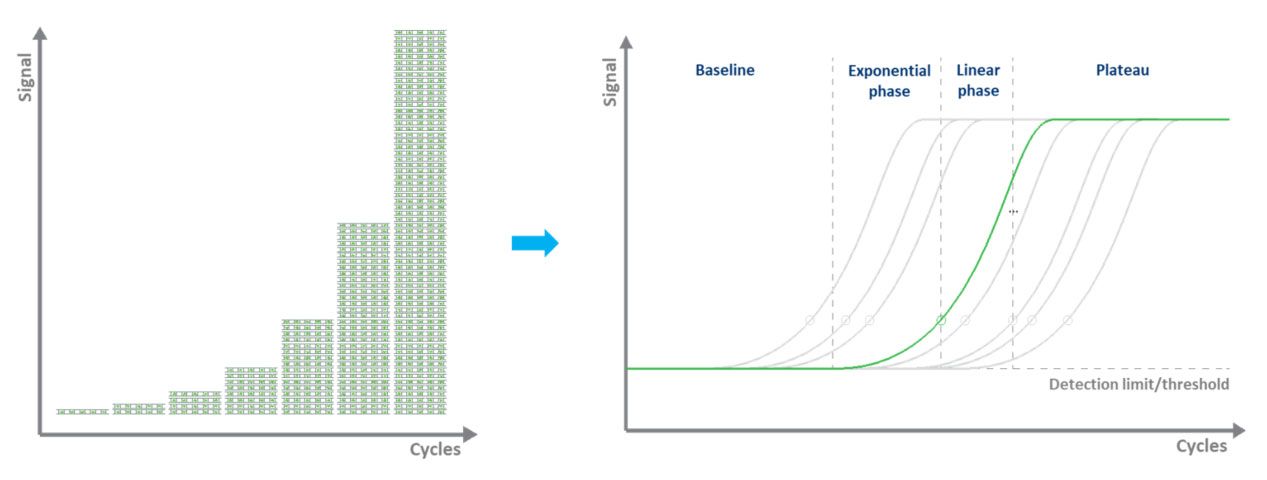
No qPCR baseado em sonda, muitos alvos podem ser detectados simultaneamente em cada amostra, mas isso requer otimização e design de sonda(s) específica(s) do alvo usadas além dos primers.Vários tipos de designs de sonda estão disponíveis, mas o tipo mais comum é uma sonda de hidrólise, que incorpora um fluoróforo e um supressor.A transferência de energia de ressonância de fluorescência (FRET) evita a emissão do fluoróforo através do supressor enquanto a sonda está intacta.Contudo, durante a reacção de PCR, a sonda é hidrolisada durante a extensão do iniciador e a amplificação da sequência específica à qual está ligada.A clivagem da sonda separa o fluoróforo do supressor e resulta num aumento da fluorescência dependente da amplificação (Fig. 4).Assim, o sinal de fluorescência de uma reação qPCR baseada em sonda é proporcional à quantidade da sequência alvo da sonda presente na amostra.Como o qPCR baseado em sonda é mais específico do que o qPCR baseado em corante, muitas vezes é a tecnologia usada em ensaios de diagnóstico baseados em qPCR.
| |
| Figura 4.Diferenças entre qPCR baseado em corante e baseado em sonda. |
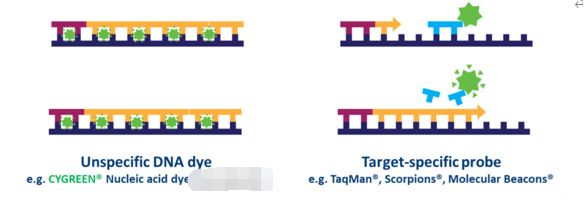
Amplificação Isotérmica
As técnicas de PCR mencionadas acima requerem equipamento de termociclagem caro para aumentar e diminuir com precisão as temperaturas da câmara para as etapas de desnaturação, recozimento e extensão.Foram desenvolvidas diversas técnicas que não necessitam de dispositivos tão precisos e podem ser realizadas em simples banho-maria ou mesmo dentro das células de interesse.Essas técnicas são chamadas coletivamente de amplificação isotérmica e funcionam com base na amplificação exponencial, linear ou em cascata.
O tipo mais conhecido de amplificação isotérmica é a amplificação isotérmica mediada por loop, ou LAMP.LAMP usa amplificação exponencial a 65⁰C para amplificar DNA ou RNA modelo.Ao realizar o LAMP, quatro a seis primers complementares às regiões do DNA alvo são usados com uma DNA polimerase para sintetizar novo DNA.Dois desses primers possuem sequências complementares que reconhecem sequências nos outros primers e as ligam, permitindo a formação de uma estrutura em “alça” no DNA recém-sintetizado que auxilia no recozimento dos primers em rodadas subsequentes de amplificação.LAMP pode ser visualizado por vários métodos, incluindo fluorescência, eletroforese em gel de agarose ou colorimetria.A facilidade de visualização e detecção da presença ou ausência do produto por colorimetria e a falta de equipamentos caros necessários tornaram o LAMP uma opção adequada para testes de SARS-CoV-2 em áreas onde os testes de laboratório clínico não estavam prontamente disponíveis, ou armazenamento e transporte de amostras não era viável, ou em laboratórios que anteriormente não possuíam equipamento de termociclagem PCR.
Horário da postagem: 19 de agosto de 2023



